Que sont les prions ? Partie I : les maladies à prions, découverte et explications

TRIBUNE Santé - C’est un petit article de FranceInfo « Élevage : les farines animales de nouveau autorisées par la Commission européenne », publié le 24/08/2021, associé à un court film de deux minutes, qui m’ont décidé à écrire cet article. Dans cet article, FranceInfo concluait :
« Auparavant, ces farines animales pouvaient être fabriquées à partir d’animaux malades ou morts, d’os, de poils ou de cerveaux. Désormais, l’Europe l’assure, les protéines animales transformées viendront uniquement d’animaux en bonne santé, destinés à l’alimentation humaine. »
La fin de la vidéo n’était pas vraiment rassurante : « Contacté, le ministère de l’agriculture n’a pas souhaité répondre ».
Les farines animales
Donc, 20 ans après l'interdiction européenne de toutes les farines animales destinées à l'élevage - interdiction liée à la crise de la "vache folle", il sera de nouveau possible de nourrir porcs et volailles avec des « protéines animales transformées » ou PAT, dans cette même Union européenne. Pour nous convaincre du bien-fondé de cette décison, on nous explique que les PAT d'aujourd'hui seraient équivalentes à de l’eau pure alors que les anciennes farines animales seraient comparables aux eaux usées…
Le texte a été publié le 18 août 2021 au Journal officiel de l'UE et devait entrer en application vingt jours plus tard, soit le mardi 7 septembre. Bruxelles a mis en avant le fait que ces produits, contenant 50 à 60 % de protéines, peuvent se substituer partiellement à l'importation très onéreuse de protéines végétales, comme le soja brésilien, qui n'en contient que 45 %.
C’est bien connu, les poissons ne font pas de vagues… Les poissons d'élevage et les animaux de compagnie pouvaient déjà être nourris avec ces farines depuis 2013 dans l’hexagone, sous la pression de Bruxelles. La Commission européenne avait édicté le règlement 56/2013, qui permettait d’utiliser des farines animales de volailles ou de porc pour nourrir les poissons d’élevage. La France et l’Allemagne, seules, avaient alors voté contre, « avant de s’incliner en renâclant ». C’était clair : si les poissons pouvaient gober de la poule et du porc, impossible pour eux de picorer d’autres poissons réduits en farine…
Désormais, il sera possible d’utiliser les farines animales PAT pour nourrir volailles et porcs avec des restrictions : du poulet pour les cochons, mais pas du cochon pour les cochons. Par contre, l'interdiction reste valable pour les ruminants herbivores comme les bovins, les chèvres et les moutons.
La France, comme l’Irlande, s'est abstenue lors du vote, et la situation reste en suspens. Le ministère de l'Agriculture a indiqué à l'AFP le 3 septembre 2021 qu'il demandait un nouvel avis à l'agence sanitaire nationale pour "se positionner" sur le sujet. Je n’en sais pas plus au moment où j’écris cet article.
La maladie de la "vache folle"
Les carcasses de bovins malades, infectés ou morts étaient, avant 2001, broyées pour nourrir des élevages de la même espèce. Cela a conduit à la propagation de l'encéphalopathie spongiforme bovine (ESB) ou maladie de la « vache folle », une maladie à prion qui s’est transmise aux consommateurs sous la forme d’un variant humain toujours mortel : la maladie de Creutzfeldt-Jacob. Le 28 février 1991, c’était la découverte du premier cas français d’encéphalopathie spongiforme bovine.
Qui empêchera une entreprise désireuse de "faire du pognon" de s'affranchir des limites imposées, s'interroge Nicolas Girodle, le porte-parole du syndicat Confédération Paysanne. "Qu'est-ce qui a provoqué la vache folle ? C'est la recherche de profit, de volume, de productivité." Comment faire confiance ? Bannies, parties à grand bruit, ces farines animales reviennent en catimini. Il est vrai que le mardi 14 novembre 2000, le Premier ministre d’alors, Lionel Jospin, avait annoncé “une suspension temporaire et générale des farines animales".
Plus d'infos à ce sujet : reportage britannique de 1990
Fraude à la viande de cheval
Comment ne pas se souvenir du scandale de « la fraude à la viande de cheval » qui éclata le 16 janvier 2013 dans les îles britanniques ? La révélation publique de la découverte de traces d'ADN équin et de viande chevaline, jusqu'à 29 %, dans les steaks hachés étiquetés au bœuf. L’affaire remonte au 30 novembre 2012, quand la Food Safety Authority of Ireland (FSAI) reçoit les résultats d'analyse du laboratoire IdentiGEN sur des échantillons achetés du 7 au 9 novembre 2012 : des salamis, des plats préparés à base de bœuf et des hamburgers au bœuf. Le test quantitatif révèle que 23 (85 %) des hamburgers de bœuf sont positifs pour l'ADN de viande porcine et 10 (37 %) pour l'ADN de cheval. Toute la chaîne du commerce alimentaire européen s’est trouvée entachée, dont des abattoirs, des intermédiaires (Draap Trading), des négociants (Spanghero) et des transformateurs (Comigel)… sans parler des supermarchés Tesco, Lidl et Iceland, ainsi que des fameuses lasagnes de Findus. Cette fraude, commise à l'échelle d’un continent, qui faisait passer de la viande de cheval, moins chère, pour de la viande de bœuf, a porté sur environ 4,5 millions de plats préparés… Imaginez le nombre de consommateurs concernés ! Il semble bien que de telles irrégularités persistent.
Alors oui, comment faire confiance ? Qui nous dit qu’une bête trouvée morte dans un champ, ou malade à son arrivée à l’abattoir, sera vraiment rejetée pour la constitution de farines animales ?
Comment faire confiance ? Petit historique :
1980 : Viande provenant d'animaux traités aux hormones (« bœuf aux hormones »), pratique qui sera interdite dans l'Union européenne fin 1980, mais restera autorisée aux États-Unis.
1981 : Le scandale de l'huile frelatée en Espagne qui a causé 20 688 victimes (entre 370 à 835 morts, suivant les décomptes). Dr Juan Tabuenca Oliver, directeur de l'hôpital pour enfants Niño Jesus, annonçait avoir identifié la cause de l'épidémie : les 210 enfants traités dans son service ont tous consommé de l’huile de colza. L'empoisonnement était dû aux composés toxiques introduits dans le processus d'affinage pour extraire l'aniline et dénaturer les huiles en vue de leur utilisation par l'industrie. Cette huile a été vendue comme étant une huile d’olive.
Crise de la dioxine en mai 1999 : du polychlorodibenzo-p-dioxine est trouvé dans des farines animales en Belgique, farines utilisées pour l'alimentation des poulets. Des dizaines de milliers de malades et abattage de sept millions de poulets et 60 000 porcs…
2008, scandale du lait de vache frelaté en Chine : certains lots de lait de consommation courante et de lait infantile produits en Chine ont contenu pendant dix mois de la mélamine = cyanuramide = cyanurotriamine, une sorte de résine toxique, afin de les faire apparaître plus riches en protéines.
2008, scandale des fromages italiens avariés ou périmés : La Republica révèle qu'une quarantaine de grandes entreprises laitières (italiennes, mais aussi britanniques, allemandes et autrichiennes) se sont débarrassées, au lieu de les détruire, de onze mille tonnes de fromage avarié ou périmé en les « recyclant ». En fait, en les intégrant à de nouveaux produits fromagers, produits vendus dans toute l'Europe… D’après l’enquête, « certains fromages pourris contenaient des vers, des crottes de souris, des bouts de fer, ou même des résidus du plastique les ayant emballés, et l'encre des étiquettes ».
2013, les tartes aux amandes et au chocolat des magasins Ikea contaminées avec de la matière fécale : « Le géant suédois de l'ameublement Ikea annonce qu'il retire de ses cafétérias dans 23 pays, des tartes sur lesquelles les autorités sanitaires chinoises ont trouvé des bactéries généralement témoins d'une contamination fécale. » Un niveau excessif de bactéries coliformes…
2017, le scandale des œufs contaminés au fipronil, un antiparasitaire, en Europe et en Asie : Une entreprise néerlandaise, Chickfriend, a revendu un traitement acaricide (contre le pou rouge des poules, en fait un acarien Dermanyssus gallinae) acheté chez Poultry-Vision (une entreprise belge), traitement destiné à des poules pondeuses. Le fipronil était utilisé par des entreprises de désinfection dans des exploitations agricoles (Pays-Bas, Belgique, Allemagne) mais ce traitement, était interdit pour les animaux destinés à la consommation humaine. Plus de 6 000 litres de produit interdit ont été saisis chez Poultry-Vision. "Des millions d'œufs de poule ont été retirés du commerce, dont des œufs estampillés « bio » provenant des Pays-Bas".
Comment ne pas être sceptique... Jean de la Fontaine écrivait dans "Le chat et un vieux rat" :
"C’était bien dit à lui ; j’approuve sa prudence :
Il était expérimenté,
Et savait que la méfiance
Est mère de la sûreté."
I - La maladie de la vache folle, ou encéphalopathie spongiforme bovine (ESB), en détails :
C’est une maladie dégénérative du cerveau, de la moelle épinière et des ganglions rachidiens des bovins. Elle est apparue au Royaume-Uni en 1986. On collectait depuis des décennies les produits d’équarrissage : graisses, os et autres déchets d’abattoirs, de bovins et d’ovins, mais également les carcasses d’animaux morts dans les fermes, afin de produire ces « farines animales » ou FVO = farine de viande et d’os.
Mais, en 1981, le procédé de production des farines animales, par pur souci de rentabilité, est modifié au Royaume-Uni. La température de cuisson est abaissée à 90°C et la pression atmosphérique utilisée diminuée, en plus de quoi l'étape d'extraction des graisses par solvants est éliminée. Cette méthode présente l’avantage d’obtenir un taux de protéines plus important dans ces farines et de réduire le coût énergétique.
Les importations de farines et d’animaux vivants ont fait entrer la maladie en France, où un premier cas est découvert début 1991.
Le 13 mars 1996, coup de tonnerre, le gouvernement britannique reconnaît l'existence d'un « lien » possible entre la maladie bovine et l'apparition de cas d'une nouvelle forme d'une maladie humaine déjà connue, la maladie de Creutzfeldt-Jakob (MCJ) dite « sporadique », du nom des deux neurologues qui l'ont décrite presque simultanément en 1920 et 1921. Cette nouvelle forme reçoit l’acronyme noté nvMCJ.
En effet, deux Britanniques habitant le nord de Londres sont décédés d'une maladie qui semblait être, à première vue, la maladie de Creutzfeldt-Jakob. Stephen Churchill et Nina Sinnott, âgés respectivement de 19 et 25 ans, sont anormalement jeunes pour développer cette maladie qui affecte exclusivement les personnes âgées. Une dizaine de cas sont recensés jusqu'à ce que le nombre atteigne 180. L’inoculation expérimentale à des macaques entraîna une pathologie présentant de nombreux points communs avec la nvMCJ, dont des lésions cérébrales similaires.
Après l'ingestion du prion, celui-ci pénètre dans les formations lymphoïdes des intestins, notamment les plaques de Peyer, dans lesquelles il peut se répliquer, puis rejoint le système nerveux.
Au total, plus de 184 624 cas d’ESB ont été officiellement confirmés au Royaume-Uni et 231 personnes recensées ont été victimes de nvMCJ (dont 178 au Royaume-Uni, 27 en France, cinq en Espagne, quatre en Irlande et aux États-Unis, trois aux Pays-Bas et en Italie, deux au Portugal et au Canada, un au Japon, à Taïwan et en Arabie saoudite).
Le passage du bovin à l'homme se fait par ingestion de cervelle et de viande. La maladie a même atteint un chat domestique (probablement contaminé par la nourriture, les aliments pour chats étant très souvent fabriqués à partir d'abats de bovins) ainsi que d’autres carnivores : lions, tigres, pumas, guépards dans les zoos britanniques. L’ESB a contaminé des antilopes et des bovidés sauvages comme le bison.
La barrière de transmission varie selon les espèces, « celles-ci étant plus ou moins permissives aux différents prions selon la séquence primaire » de leur propre protéine. « Le degré de similitude de la séquence primaire (du prion) inoculée et la protéine (correspondante) de l’hôte est un élément clé de l’efficacité de transmission inter-espèce (Prusiner et al., 1990). » [...] « Ainsi des espèces comme le lapin, le chien, le porc et le cheval, semblent être résistantes à la plupart des prions issus d’espèces différentes, contrairement aux hamsters ou aux campagnols qui sont très permissifs. » selon Angélique Igel-Egalon.
On a expérimentalement pu constater la transmission possible à d'autres mammifères (la souris, le porc et certains singes), en recourant à des méthodes particulièrement invasives (injection dans le cerveau).
Il existe différentes ESST (encéphalopathies subaiguës spongiformes transmissibles) qui touchent d'autres ruminants, sans qu'aucun lien direct ne soit prouvé avec l'ESB. Ces maladies n’existent pas chez les poissons et les oiseaux. Le chien n’est pas non plus concerné par ce type de maladies neurologiques.
À titre indicatif, un cerveau de souris contaminé contient suffisamment d’infectiosité pour transmettre la maladie à un million de congénères !
En France, le réseau d’épidémio-surveillance clinique des bovins a permis le diagnostic de 102 cas d’ESB en 2000, 91 en 2001 (dont 83 cas à l’abattoir soit 0,034 cas pour mille bovins abattus), 41 en 2002, 13 en 2003, huit en 2004 et seulement deux en 2005. En 2008, un seul cas a été détecté en abattoir.
« La mesure de sécurité la plus importante est le retrait systématique de la consommation, de tous les produits qui pourraient être porteurs de l’agent infectieux, et ce, pour chaque ruminant abattu. » Ces produits éliminés sont dénommés les « Matériels à Risque Spécifiés » ou MRS. Ils sont systématiquement incinérés dans le cadre de la réglementation française. « … des organes comportant des tissus lymphoïdes (amygdales, intestins) sont considérés comme des MRS et sont retirés quel que soit l’âge de l’animal. »
« Parallèlement, selon les estimations, 100 000 tonnes d’anciennes denrées alimentaires contenant du collagène ou de la gélatine de ruminants sont éliminées chaque année dans l’Union, car, en vertu des règles actuelles relatives à l’interdiction des farines animales, elles ne peuvent pas être utilisées dans l’alimentation des animaux d’élevage » [...] « Il y a donc lieu d’abroger l’interdiction d’utiliser du collagène et de la gélatine de ruminants dans l’alimentation des animaux d’élevage non ruminants. »
Chez les animaux malades, les symptômes n’apparaissent pas avant l’âge de deux ans et en général quatre à cinq ans après la contamination, surtout sur des bêtes de cinq à sept ans. L’animal s’isole du troupeau. La production laitière d’une vache atteinte d’EBS décroît tandis que l’évolution de la maladie débouche sur des troubles du comportement (nervosité, agressivité), hyperexcitabilité (au bruit, au toucher, à la lumière), tremblements, troubles de la locomotion avec peine à tenir debout puis chute sans parvenir à se relever. L'évolution peut durer d'une semaine à un an et se termine toujours par le décès.
II - Qu’est-ce qu’un prion ?
La théorie la plus généralement admise de la cause de la maladie de la « vache folle » est celle des prions (« PRotinaceous Infectious Only »), même si certains chercheurs continuent à travailler des hypothèses virales et bactériennes.
II A - Un prion n’est pas un virus :
En 1953, André Lwoff a proposé une définition des virus en quatre points, qui montre que les prions, comme nous allons le voir, sont très différents :
1- Un virus n'a ni cytoplasme ni noyau, mais est porteur d’acide nucléique et de protéines en une structure définie et constante, qui possède des éléments de symétrie.
2- Un virus ne renferme qu'un type d'acide nucléique, ADN ou ARN, jamais les deux.
3- Un virus est incapable de se diviser. Il se reproduit uniquement, à partir de son matériel génétique (l'acide nucléique).
4- Un virus est un parasite absolu, il possède l'information nécessaire à la synthèse de ses propres constituants, mais n'a pas les moyens d'exprimer cette information. Cela signifie qu’il nécessite un hôte.
Tous ceci reste globalement exact.
On pourrait compléter en précisant qu’un virus passe par deux phases :
1. Une phase extracellulaire sous la forme d’une particule virale infectieuse : comprenant un acide nucléique, souvent englobé dans une capside de protéines ;
2. Une phase intracellulaire soit sous forme dormante, soit détournant la machinerie cellulaire au profit de sa propre réplication « en parasitant tout ou partie du métabolisme de son hôte. »
II B - Si ce n’est pas un virus… qu'est-ce qu'un prion ?
Un prion est, au sens restreint, un agent pathogène constitué d'une protéine (de 253 acides aminés pour le « prion au sens strict ») glycosylée, dont la conformation ou le repliement est anormal, et qui est susceptible d’entraîner des maladies neurodégénératives diverses. Le « prion » dérive donc d'une glycoprotéine qui existe à l'état naturel chez tous les mammifères et chez l'Homme. Cette protéine ne provoque aucune réaction de type immunitaire de la part de l'organisme, pas de formation d'anticorps. L'hypothèse serait que cette protéine changerait de forme pour une cause inconnue, et qu'elle deviendrait alors pathologique.
La protéine modifiée pénètre dans la cellule par endocytose (processus par lequel la membrane d'une cellule enveloppe et absorbe une particule) ou par le biais du récepteur spécifique du "prion normal", la protéine LRP (Laminin Receptor Protein).
Une fois la protéine anormale entrée dans la cellule, elle transforme les "prions normaux" en prions résistants. Les protéases n'étant plus capables de les détruire, ces protéines s'accumulent et ainsi la formation de plaques amyloïdes pour finir par provoquer la mort du neurone.
Pour résumer : les prions infectieux sont des « agents transmissibles non conventionnels (ATNC) » ou « prions pathogènes » issus d’une protéine normale par mauvais repliement ou par mutation. La glycoprotéine normale ubiquitaire ou PrPC (abréviation de protéine prion cellulaire, l'isoforme normale), sous une conformation anormale (ou mal repliée) est alors dénommée PrPSc (abréviation de protéine prion de la scrapie).
Ces modifications conformationnelles rendent la protéine anormale insoluble et résistante aux agents protéolytiques, à la congélation, à la dessication et à la chaleur aux températures normales de cuisson. Le prion n'est détruit que par une température très élevée (133°C pendant 20min sous une pression de trois atmosphères). Les prions sont très résistants à l'irradiation UV, contrairement aux acides nucléiques.
À l’inverse des agents infectieux tels que les virus ou les bactéries, ou encore des parasites, il ne dispose pas d'acide nucléique (ADN ou ARN) comme support de l'information infectieuse. Il peut néanmoins parfois s’associer à un acide nucléique.
Les prions de mammifères sont les agents responsables des encéphalopathies spongiformes transmissibles (= EST) ou maladies à prion. Ces maladies sont caractérisées par une longue période d'incubation, en général plusieurs années, jusqu'à 40 ans chez l'Homme. Les prions ne sont pas spécifiques des mammifères puisqu’on en a retrouvés chez les champignons comme chez Saccharomyces cerevisiae (levure de boulanger) sans qu’intervienne ici une notion pathogène.
Il existe des formes héréditaires, des formes alimentaires et des formes iatrogènes (hormone de croissance contaminée, électrodes, greffes de dure-mère issue de cadavres). Une voie de contamination mère-veau est aussi soupçonnée.
La maladie de la vache folle (ESB), sous sa forme classique, était donc liée à l'ingestion par les bovins, de farines animales contaminées qui étaient utilisées dans leur alimentation. Chez l'Homme, la variante de la maladie de Creutzfeldt-Jakob est causée par l'agent de l'ESB transmis par la consommation de tissus, en particulier nerveux mais aussi de viande, d’animaux atteints.
L’origine iatrogène est, elle, en particulier due à des injections d’hormone de croissance préparée à partir de tissus humains contaminés par une protéine prion anormale (PrPSc).
L’hormone de croissance naturelle (= somatotropine = growth hormone = GH) est un polypeptide de 191 aminoacides sécrété par les cellules somatotropes de l'hypophyse antérieure selon un régime pulsatile, sous le contrôle de l’hypothalamus. On sait qu’un manque de GH pendant l'enfance provoque un nanisme harmonieux. Le traitement comprend une hormonothérapie de substitution. Initialement, elle faisait appel à une hormone de croissance humaine dite « extractive » à partir d'hypophyses prélevées sur des cadavres…
En France, le prélèvement s’effectuait par le nez sans ouvrir la boite crânienne. L’affaire se déroule entre 1982 et 1985… Des lots, non traités à l'urée, contenant tous de la poudre d'hormone de croissance, ont été distribués après le 15 mai 1985 à 800 enfants. Ces lots ont été causes d’une maladie à prion (maladie de Creutzfeldt-Jakob = MCJ).
Dès 1979-1980, alors que les prions n'avaient pas encore été découverts, le professeur Montagnier demandait qu’« une attention particulière soit portée au danger de transmission de la MCJ » et préconisait, faute de certitudes, « d'écarter au moins certains donneurs d'hypophyse "à risque" ». Malgré ces alertes de notre prix Nobel, l’avertissement n’a pas été écouté. Oui, quand notre prix Nobel alerte il n’est pas entendu, encore moins écouté... Cela nous renvoie à une situation actuelle.
57 % des cas de MCJ transmis par l'hormone de croissance ont été enregistrés en France. En 1983, 17 000 glandes bulgares avaient été importées. Corruption, vol de documents et 120 enfants décédés. Le scandale n’éclatera qu’en 1991.
« Après vingt-quatre ans, deux procès et une relaxe générale, la Cour d’appel de Paris a relaxé au civil, lundi 25 janvier 2016, le professeur Fernand Dray et l’ancienne pédiatre Elisabeth Mugnier, les deux derniers prévenus en vie dans l’affaire dite de « l’hormone de croissance ». Maitre Bernard Fau, s’exprimant au nom des 21 parties civiles avait alors dénoncé un « naufrage » et l’« incapacité de la justice française à appréhender ce type de grand scandale sanitaire ». « La justice a estimé que la responsabilité civile de Fernand Dray et Elisabeth Mugnier n’avait pas été engagée dans ce scandale sanitaire. Les victimes ne seront pas indemnisées. »
Un quart de siècle plus tard, il en sera probablement de même avec les quatre thérapies géniques (Comirnaty® de Pfizer, Spikevax® de Moderna et les 2 OGM Vaxzevria® d’AstraZeneca et Janssen® de Johnson&Johnson) injectées à nos anciens et enfants. Anciens non protégés car ne produisant pas assez d’anticorps et enfants qui ne contractent qu’une covid-19 non pathogène pour eux… La pathogénie de la spike (sur cellules endothéliales, cardiaques et nerveuses) sera finalement reconnue. Messieurs Macron et Véran auront alors respectivement de l’ordre de 70 et 66 ans quand ils viendront à la barre se présenter comme de doux agneaux innocents… si tout se passe bien.
Pour les maladies à prions, il n'existe à l'heure actuelle ni traitement ni aucun vaccin.
II C - Fonction de la protéine prion et conséquences de son changement de forme :
L’ensemble de ces maladies se caractérise donc par une dégénérescence du système nerveux central (cerveau, moelle épinière, ganglions rachidiens) liée à la propagation ou multiplication de prions chez l’hôte infecté. D'un point de vue anatomopathologique, on observe au niveau de l'encéphale la formation de vacuoles (donnant un aspect spongieux au cerveau, d'où le terme de « spongiforme »), une mort des neurones, une gliose (multiplication des astrocytes et de la microglie)... et l'accumulation d'une protéine de l'hôte que l’on peut considérer comme un amas pathogène se présentant sous formes de plaques amyloïdes caractéristiques (appelées ainsi car elles évoquent de l’amidon). Amas extracellulaires, quasi exclusivement constitué de prions et plus ou moins importants selon les pathologies.
En 1991, le groupe de Stanley Prusiner établit que la PrP n'est pas une protéine codée par le génome d’un l'agent infectieux mais bien par le génome de l'hôte. La protéine PrPC normale est présente essentiellement dans le système nerveux (neurones et glie du cerveau et la moelle épinière) à la surface des cellules mais aussi sur d’autres cellules non cérébrales, telles que des cellules immunitaires et des cellules épithéliales et endothéliales. Cette protéine est monomérique et ancrée à la surface externe des cellules. La PrPC ne possède pas de domaine transmembranaire.
Elle a de multiples fonctions qui restent à affiner :
- Elle est impliquée dans le développement du système nerveux chez l'embryon du fait notamment de sa contribution à l'adhérence cellulaire (voir point suivant).
- Également dans les processus de différenciation et d’adhésion des cellules entre elles et dans l’intégrité des barrière épithéliales et endothéliales constituant les barrières physiologiques de l'organisme : barrière intestinale et barrière vasculaire (migration cellulaire, voir point suivant) et barrière hémato-encéphalique. À noter que la localisation préférentielle de la PrPC se situe au niveau des jonctions intercellulaires.
La protéine normale PrPC (dans son domaine dit globulaire des résidus ̴125-230 seul ici représenté) comporte trois hélices alpha (en brun-violet) et deux feuillets béta en bleu. Elle est soluble et sensible aux protéases. La protéine anormale PrPSc (associée ici à la protéine normale) est riche en feuillets béta (en bleu). Elle est insoluble, résistante aux protéases et autoréplicative. En s’agrégeant, les protéines PrPSc donnent naissance à des structures quaternaires : les « fibres amyloïdes ».
Auteur schéma : Dr. Human Rezaei.
 La protéine normale PrPC (dans son domaine dit globulaire des résidus ̴125-230 seul ici représenté) comporte trois hélices alpha (en brun-violet) et deux feuillets béta en bleu. Elle est soluble et sensible aux protéases. La protéine anormale PrPSc (associée ici à la protéine normale) est riche en feuillets béta (en bleu). Elle est insoluble, résistante aux protéases et autoréplicative. En s’agrégeant, les protéines PrPSc donnent naissance à des structures quaternaires : les « fibres amyloïdes ».
La protéine normale PrPC (dans son domaine dit globulaire des résidus ̴125-230 seul ici représenté) comporte trois hélices alpha (en brun-violet) et deux feuillets béta en bleu. Elle est soluble et sensible aux protéases. La protéine anormale PrPSc (associée ici à la protéine normale) est riche en feuillets béta (en bleu). Elle est insoluble, résistante aux protéases et autoréplicative. En s’agrégeant, les protéines PrPSc donnent naissance à des structures quaternaires : les « fibres amyloïdes ».- La protéine est présente sur les monocytes, les cellules dendritiques et les lymphocytes T. Elle semble jouer un rôle dans la migration trans-endothéliale des monocytes et son expression est augmentée au cours de l'activation des lymphocytes T. La PrPC participe à interaction entre les cellules dendritiques et les lymphocytes T.
- Elle aurait aussi un rôle dans la survie cellulaire (protecteur antioxydante lors d’un stress oxydatif et vis-à-vis de la mort cellulaire programmée ou apoptose [11]. Des études réalisées sur des coupes tissulaires ont révélé que la liaison de la PrPC par des anticorps spécifiques induit l'apoptose des neurones du cervelet et de l'hippocampe. L’hippocampe est une structure bilatérale du cerveau, primordiale pour la mémoire à court terme. Des neurones en culture provenant de souris chez lesquelles le gène codant pour la PrPC a été invalidé (souris PrP −/−) sont plus sensibles à un stress oxydant induit par des dérivés réactifs de l'oxygène ou DRO (radicaux libres, des ions oxygénés et peroxydes) [14].
- Elle jouerait un rôle dans l’homéostasie du cuivre : capacité de lier le cuivre sous forme d'ion cuivrique (Cu ++), et plus faiblement le zinc (Zn ++) et le manganèse (Mn ++) suggérant une contribution de la PrPC à la protection contre des concentrations toxiques en ions métalliques.
Structure complète de la protéine normale PrPC avec sa partie globulaire en bas et sa partie terminale, longue chaîne polypeptidique non structurée capable de lier les ions Cu ++
 Structure complète de la protéine normale PrPC avec sa partie globulaire en bas et sa partie terminale, longue chaîne polypeptidique non structurée capable de lier les ions Cu ++
Structure complète de la protéine normale PrPC avec sa partie globulaire en bas et sa partie terminale, longue chaîne polypeptidique non structurée capable de lier les ions Cu ++- Cette protéine aurait également un rôle dans le repliement d’autres protéines.
- In vivo, la PrPC semble jouer un rôle dans des réponses comportementales perturbées dans les maladies à prion : la régulation du sommeil, l'apprentissage et la persistance de la mémoire.
- D’autres pathologies cérébrales (ischémie, gliome) et plusieurs maladies neuromusculaires (dénervation neurogène, myosite à inclusions, polymyosite) s’accompagnent de la surexpression de la PrPC.
[11] Kim BH, Lee HG, Choi JK et al. La protéine prion cellulaire (PrP c) prévient la mort cellulaire neuronale par apoptose et le dysfonctionnement mitochondrial induit par la privation de sérum. Brain Res Mol Brain Res 2004; 124 : 40-50. [Google Scholar]
[14] Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA. Les cellules déficientes en protéines prions présentent une réponse altérée au stress oxydatif en raison d'une diminution de l'activité SOD-1. Exp Neurol 1997; 146 : 104-12.
Caractéristiques neuropathologiques de la maladie de Creutzfeldt-Jakob
« Coupes histologiques et immunohistologiques de cortex frontal provenant d’un patient décédé sans syndrome neurologique (contrôle), ou d’un patient atteint de la maladie de Creutzfeldt-Jakob (CJD). » Les sections de cerveaux sont colorées avec l’hématoxyline-éosine (HE, colonne de gauche – l’hématoxyline colore les noyaux en bleu-violet, l’éosine révèle le cytoplasme en rose), ou marquées avec un anticorps reconnaissant la protéine GFAP (Glial fibrillary acidic protein) des cellules gliales de type astrocyte notamment (colonne du milieu), ou marquées avec un anticorps reconnaissant la protéine PrP (colonne de droite). GFAP est codée par le bras long du chromosome 17 (17q21) et participe au fonctionnement de la barrière hémato-encéphalique. « Chez le patient malade, la perte neuronale et la spongiose sont visibles à la coloration HE (taches blanches). Une forte prolifération des cellules gliales (nommée gliose) et des dépôts de PrP sont détectés respectivement par le marquage des anticorps anti-GFAP et anti-PrP des sections de cortex de patient atteints de MCJ. » Par Angélique Igel-Egalon.
 Caractéristiques neuropathologiques de la maladie de Creutzfeldt-Jakob
Caractéristiques neuropathologiques de la maladie de Creutzfeldt-JakobII D - Selon la mutation, la pathologie est différente :
Le prix Nobel de médecine 1997, Stanley Prusiner, considérait que des protéines de type prions sont à l'origine de l'ensemble des maladies neurodégénératives. Les travaux récents menés sur d’autres protéinopathies (maladies d’Alzheimer et de Parkinson) montrent qu’elles partagent des propriétés avec les prions sur bien des aspects.

La protéine PrPC, elle-même, pourrait même être impliquée dans le diabète de type 1 ou juvénile, maladie caractérisée par une attaque par le système immunitaire des cellules produisant l'insuline (dans le pancréas). Les prions représentent donc une classe de protéines mutées pathogènes. Le gène codant la protéine prion PrP normale se trouve sur le bras court (dit « p ») du chromosome 20 à la position p13. Des mutations ou des insertions de nucléotides sur le gène se produisent en différents lieux puis s’accumulent…
Au sein de la même espèce hôte, les maladies à prion peuvent donner lieu à tout un panel de formes, « toute une panoplie de configurations » selon Angélique Igel-Egalon. Ces variations incluent :
1. Le temps d’incubation de la maladie (parfois extrêmement long),
2. Le profil lésionnel (tropisme tissulaire, distribution des dépôts de PrPSc et vacuolisation)
3. Leurs propriétés biochimiques (profil de glycosylation, degré de résistance à la protéolyse et à la dénaturation, site de coupure des protéases…)
Ces caractères sont stables et conservés aux cours des transmissions successives.
Ainsi, le syndrome de Gerstmann-Sträussler-Scheinker = GSS = SGSS (sur laquelle nous reviendrons) est une encéphalopathie spongiforme transmissible (EST) humaine, le plus souvent familiale, héréditaire, à transmission dominante. Elle affecte les patients de 30 à 60 ans et la survie varie de un à onze ans avec une moyenne de cinq ans. Elle se caractérisée parfois par un nystagmus (mouvement involontaire saccadé lent-retour rapide des yeux, orienté dans une direction donnée), des troubles de la parole (dysarthrie) et une difficulté à manger (nécessitant souvent une nutrition par gastrostomie endoscopique percutanée), parfois une surdité, une raideur musculaire, des mouvements de type Parkinson et une ataxie (= syndromes cérébelleux) avec démence tardive. Les ataxies se traduisent par des troubles de la coordination des mouvements volontaires sans faiblesse musculaire. D’une façon générale, des plaques amyloïdes siègent au niveau du cervelet mais sont aussi distribuées dans le cortex cérébral et les noyaux gris centraux.
La GSS a été associée à une variété de mutations ponctuelles ou d’insertions sur le gène PRNP codant la protéine PrP et donc dans la protéine prion elle-même, de situations variables : 102, 105, 117, 131, 145, 187, 198, 202, 212, 217, 218 et 232.
Il existe donc une hétérogénéité clinique marquée chez les patients atteints de la maladie du SGS, même parmi ceux porteurs de la même mutation. Dans plusieurs familles avec GSS, les mutations responsables sont inconnues.
- La forme la plus fréquente correspond à une mutation de la protéine prion qui se situe en position 102 de la protéine ou l’acide aminé proline est remplacé par l’acide aminé leucine. On écrira P102L. Elle est responsable de la forme dite ataxique. Cette forme se rencontre dans plusieurs pays d'Europe et au Japon. Le décès survient dans un délai de un à onze ans. À l'examen neuro-pathologique, la spongiose est sévère et il existe de nombreuses plaques amyloïdes dans le cervelet, qui sont particulières (multicentriques).
- Une mutation en position 105 de la protéine, où là encore une proline est remplacée par une leucine (P105L). Caractérisée par une démence tardive et une paraparésie spastique (= faiblesse progressive, accompagnée de contractures musculaires, dans les jambes).
- La mutation 117 a été décrite dans deux familles (allemande et alsacienne). A117V (A pour alanine et V pour valine) se traduit par une démence associée à des signes évoquant une maladie de Parkinson. Les plaques amyloïdes sont uni ou multicentriques.
- La mutation 145 est quant à elle à l'origine d'un tableau clinique proche de l'Alzheimer. Les plaques sont constituées de PrP (prion protein) tronquée du fait d’une mutation « stop » en position 145. Il existe aussi une dégénérescence neurofibrillaire simultanée comme pour 198 et 217.
- La mutation F198S a été découverte dans une grande parenté de l'Indiana (Indiana Kindred). F pour phénylalanine et S pour sérine. Coexistence de plaques multicentriques et de dégénérescences neurofibrillaires identiques à celles de l'Alzheimer avec protéine Tau hyperphosphorylée (voir maladie d’Alzheimer en IIIA).
- Mutations Q217R (une famille suédoise), Q pour glutamine et R pour arginine. Il y a également ici coexistence de plaques multicentriques et de dégénérescences neurofibrillaires identiques à celles de l'Alzheimer avec protéine Tau hyperphosphorylée (voir maladie d’Alzheimer en III A).
Lors de l'infection, l'agent prion, agent pathogène responsable de l'infection, pénètre le neurone, où pour des raisons et par un mécanisme encore mal compris il se multiplie, en dépliant/repliant les protéines PrPC en protéines PrPSc. Cette forme n'étant plus dégradée par protéolyse s’accumule dans la cellule, finit par la tuer et former des plaques de dépôts dans le cerveau.
Les maladies à prions sont transmissibles d’un individu à l'autre et dans une certaine mesure d'une espèce à l’autre. Ils peuvent persister durant plus de 15 ans, notamment dans le sol où ils restent infectieux… Quelle erreur de ne pas déclarer ses animaux d’élevage et de les enterrer dans son champ...
II E - La protéine P53 peut-elle se comporter en prion-like cause de cancers ?
Le gène p53 (ou TP53 pour « tumor protein 53 ») est situé sur le chromosome 17. Il est inactivé dans près de 50 % des cancers humains. La protéine p53 à fonction endommagée, qui en provient, a été découverte en 1979. La communauté scientifique ignorait alors que ces gènes isolés chez la souris (1983) et l’Homme (1984) étaient mutés.
L’étude des propriétés de ces gènes mutés a conduit à les classer initialement dans le groupe des oncogènes (gènes dont l’expression favorise l’apparition de cancers). En fait, il s’agit de gardiens de l'intégrité cellulaire, de gènes suppresseurs de tumeurs.
La protéine p53 est « la gardienne du génome ». Pour que la protéine P53 soit active, il lui faut former un tétramère de quatre p53 fonctionnelles. Dans une cellule normale, il n’y a que très peu de p53 du fait de sa faible demi-vie (20 minutes) et de l’action d’une enzyme Mdm2 (pour « murine double minute 2 ») qui, en se fixant sur la protéine p53, la régule négativement en favorisant sa dégradation. Mdm2 est une sorte d’« étiquette » permettant d’adresser la molécule p53 qui la porte vers un système destructeur : le protéasome. L’interaction entre les protéines p53 et Mdm2 a été découverte en 1992.
Une mutation amplificatrice de Mdm2 inactive complètement p53. Celle-ci ne pourra intervenir ou dans les réparations de l'ADN ou dans l'apoptose (un processus physiologique de mort cellulaire programmée) ce qui va donner naissance à une multiplication anarchique de cellules = un cancer (voir schéma très simplifié ci-contre emprunté à Wikipédia).
La protéine kinase ATM (en anglais ataxia telangiectasia mutated) est activée principalement à la suite d'une cassure double-brin dans l’ADN et phosphoryle Mdm2 qui d’un état de régulateur négatif passe à un état régulateur positif de p53.
 La protéine kinase ATM (en anglais ataxia telangiectasia mutated) est activée principalement à la suite d'une cassure double-brin dans l’ADN et phosphoryle Mdm2 qui d’un état de régulateur négatif passe à un état régulateur positif de p53.
La protéine kinase ATM (en anglais ataxia telangiectasia mutated) est activée principalement à la suite d'une cassure double-brin dans l’ADN et phosphoryle Mdm2 qui d’un état de régulateur négatif passe à un état régulateur positif de p53.En cas d’activation suite à une cassure de l’ADN, la protéine kinase ATM phosphoryle Mdm2 et augmente ainsi l’affinité entre Mdm2 et l’ARN messager de p53 (p53 ARNm-Mdm2) induisant la synthèse de la protéine p53. C'est le premier exemple démontrant qu’un ARN messager peut contrôler la fonction de la protéine qu’il code en interagissant avec son régulateur, ici Mdm2.
p53 est séparée de Mdm2 et va agir sur un grand nombre de gène comme « facteur de transcription ». L’ensemble de ce programme peut conduire à deux résultats : l’arrêt du cycle cellulaire suivi d’une réparation ou, si la réparation n’est pas effective, l’apoptose (autodestruction).
Les gènes RAS sont des gènes suppresseurs de tumeur très importants dans l'apparition des cancers. L’un de leur produit, la protéine ARF (ADP-ribosylation factor) s'est avérée induire un arrêt du cycle cellulaire en se liant à Mdm2 favorisant sa dégradation et conduisant ainsi à la stabilisation de p53.
Ce qui précède est ultra-simplifié, le gène TP53 étant en fait transcrit en huit ARN messagers différents pouvant être traduits en douze isoformes (formes distinctes). Chacune semble avoir une propriété différente. Près de 50 % des cancers humains ont une p53 mutée ou perturbée qui a perdu ses capacités anti prolifératives et apoptotiques. Dans les cancers du col de l’utérus liés à des virus de type papillomavirus humains (=HPV), une protéine virale (protéine E6) se fixe spécifiquement sur p53 et pour certains types de virus la détruit ce qui conduit à situation similaire à celle qu’une tumeur avec p53 mutée.
Les mutations de p53 sont dites dominantes négatives. En d’autres termes, une copie mutée domine une copie ayant conservé sa fonction. Chez un individu p53+/- (un allèle sauvage = wild type et un allèle muté), les deux types de protéines seront produites. Il s’en suivra 15 tétramères non fonctionnels contre un tétramère fonctionnel. La mutation du gène peut être liée aux rayons UV (cancers de la peau), au tabac (cancers bronchiques), à l’alcool (cancers hépatiques), à une toxine (aflatoxine) d’un champignon (Aspergillus) proliférant notamment sur des graines conservées en atmosphère chaude et humide dans les pays dits en voie de développement.
On savait que p53 de type sauvage (WT) = P53WT, pleine longueur, était capable d’agréger in vitro. De plus, des cellules en culture sont capables d’internaliser ces agrégats, qui co-agrègent ensuite avec la protéine p53 soluble, endogène à ces cellules. Elle se comporte donc comme un prion.
Une accumulation de la protéine a, par ailleurs, été observée dans des cellules tumorales, laissant penser que l’agrégation de p53 se produit également in vivo. Récemment, plusieurs études ont étendu le concept du prion à l'agrégation pathologique dans les tumeurs malignes impliquant une p53 mal repliée.
p63 (Yang et al. 1998) et p73 (Jost et al. 1997 ; Kaghad et al. 1997) existent sous forme de multiples variantes protéiques. Elles sont deux homologues structurels et fonctionnels du facteur de transcription suppresseur de tumeur p53 avec des activités similaires ou antagonistes. p63 et p73 possèdent aussi des fonctions biologiques distinctes et uniques : développement des épithéliums (p63) et développement des systèmes nerveux (p73). L'agrégation de p53 et sa coagrégation avec les membres de la famille p53 (p63 et p73) ont été montrées. Certains mutants p53 exercent un effet régulateur négatif dominant sur p53 de type sauvage (WT). La base de cet effet dominant-négatif est que le mutant de type amyloïde p53 convertit WT p53 en une espèce agrégée, conduisant à la perte de sa fonction suppresseur de tumeur.
Conclusion : le comportement de type prion des mutants p53 oncogènes fournit une explication pour ses propriétés dominantes négatives, y compris le potentiel métastatique élevé des cellules cancéreuses porteuses de mutations p53. L'agrégation de mutants p53 implique des propriétés de type prion dans l’origine de nombreux cancers. L'inhibition de l'agrégation de p53 semble représenter une cible prometteuse pour une intervention thérapeutique chez les patients atteints de tumeurs malignes.
Une approche thérapeutique consiste aussi à empêcher Mdm2 de réduire les niveaux de p53 en recherchant par exemple de nouveaux inhibiteurs d'interaction Mdm2-p53.
Pour plus d'infos : Étendre le concept du prion à la biologie du cancer : effet dominant-négatif des agrégats de suppresseur de tumeur p53 mutant. Silva JL et al. Biosci Rep. 2013. PMID : 24003888 (25/7/2013).
II F - Faut-il élargir la notion de prions aux viroïdes ?
À noter que certains auteurs pensent qu’il faudrait élargir la notion trop restrictive de prions aux viroïdes réduits à un seul brin d’ARN circulaire (et nous le verrons à certains ARN : chapitre V). Les viroïdes sont les plus petites entités biologiques connues. Un viroïde est une particule, plus petite qu’un virus et seulement composée d’ARN (acide ribonucléique) sans capside ni enveloppe protéique. Les viroïdes, simples brins d'ARN sont bouclés sur eux-mêmes.
Les ARN génomiques viroïdes sont infectieux et transmissibles entre les cellules et les organismes.
Viroïdes et éléments de type viroïde :
À la fin des années 1960, Theodor O. Diener, spécialiste américain des maladies végétales, caractérisait le premier d'une nouvelle classe d'agents pathogènes végétaux constitués par une simple molécule d'ARN (environ 300 nucléotides) dépourvu d’enveloppe et de capside et se réduisant à un molécule circulaire fermée de manière covalente. Circulaires, ces ARN n'ont pas besoin de séquence d'amorçage pour être copiés. Les viroïdes ne codent aucune protéine ce qui les différencie des virus acapsidés. Ils appartiennent cependant au règne des virus car il se fait répliquer en infectant une cellule hôte, tout en ayant des effets pathogènes comparables à ceux des virus encodant des protéines.
Certains pensent que la vie, à ses débuts, pourrait ressembler à ces molécules. Ils nomment ce monde ancien avant l'apparition de la vie sous sa forme actuelle : le monde d'ARN.
Les viroïdes affectent les cultures comme les tomates, les pommes de terre, des arbres fruitiers et des cocotiers. On ne connait pas de maladie humaine à une exception près, mais ils peuvent anéantir les cultures, et provoquer de sérieuses pertes économiques.
 PSTVd = Potato Spindel Tuber Viroid. Symptômes sur récolte : réduction de la taille des tubercules avec un allongement en fuseau et bosselures au niveau des yeux… Le viroïde de la maladie des tubercules en fuseau (PSTVd, Potato spindle tuber viroid) de la famille des Pospiviroidae est l'espèce-type. C'est l'agent d'une maladie qui affecte diverses espèces de la famille des Solanaceae, principalement la pomme de terre, mais aussi la tomate et le tabac. C'est le premier viroïde qui a été identifié. Il est constitué d'une petite molécule circulaire d'ARN, étroitement apparentée à celle du viroïde du rabougrissement du chrysanthème (CSVd, Chrysanthemum stunt viroïd). Il est présent en Amérique et en Europe.
PSTVd = Potato Spindel Tuber Viroid. Symptômes sur récolte : réduction de la taille des tubercules avec un allongement en fuseau et bosselures au niveau des yeux… Le viroïde de la maladie des tubercules en fuseau (PSTVd, Potato spindle tuber viroid) de la famille des Pospiviroidae est l'espèce-type. C'est l'agent d'une maladie qui affecte diverses espèces de la famille des Solanaceae, principalement la pomme de terre, mais aussi la tomate et le tabac. C'est le premier viroïde qui a été identifié. Il est constitué d'une petite molécule circulaire d'ARN, étroitement apparentée à celle du viroïde du rabougrissement du chrysanthème (CSVd, Chrysanthemum stunt viroïd). Il est présent en Amérique et en Europe.L'ARN viroïdal contamine les plantes via leur système de vascularisation et est transmis par reproduction végétale, lors de contacts entre plantes blessées, ou par les insectes.
Depuis cette découverte fondamentale, environ 45 espèces de viroïdes différentes ont été décrites. Certains restent latent et d’autres provoquent le dépérissement de la plante. Ils ont été classés en deux familles taxonomiques selon le mode de reproduction. La forme circulaire mature des viroïdes ressemble superficiellement à la structure secondaire de la forme S1 du « prion viennois » qui sera décrit ci-dessous. Elle est infectieuse et transmissible entre les cellules et les organismes.
Les membres des plus grands (famille Pospiviroidae) sont connus pour infecter d'importantes cultures économiques, telles que la tomate (Solanum lycopersicum) et la pomme de terre (Solanum tuberosum). Les symptômes varient selon la souche viroïde, la variété végétale et les conditions climatiques ; mais sont principalement caractérisés par une croissance réduite et une chlorose des feuilles (Flores et al 2005).
Les membres plus petits de la famille des Avsunviroidae regroupent trois genres et quatre espèces de viroïdes à réplication, s’effectuant dans les chloroplastes (note 1) des plantes. Ils se répliquent par un mécanisme de cercle roulant symétrique faisant des séquences ARN appelées ribozymes « en tête de marteau ». Pour expliciter les mécanismes de reproduction des viroïdes, sans entrer dans les détails et provoquer des inflammations cérébrales concomitantes inutiles, il est possible de faire appel au schéma suivant : la réplication se fait par un mécanisme de « cercle roulant ».

Note 1 : Le chloroplaste est un organite spécifique aux cellules végétales vertes où se déroule la photosynthèse. Le chloroplaste est probablement issu une fusion (endosymbiose) d'une cyanobactérie (micro-organisme qui isolé peut proliférer de manière massive et rapide entraînant un changement de couleur de l’eau - rouge, vert... et une odeur nauséabonde) par une cellule eucaryote primitive. Le chloroplaste est séparé du cytoplasme par une double membrane.
 Chloroplastes : © Kristian Peters
Chloroplastes : © Kristian PetersLe "virus" de l'hépatite D = delta, spécifique de l'Homme, est un virus qui ne peut se multiplier qu'en présence du virus de l'hépatite B (virus auxiliaire = virus assistant) dont il "emprunte" l'enveloppe. Le virus d'hépatite Delta est donc proche des viroïdes mais il s’en distingue par le fait qu’il n’utilise pas les enzymes de la cellule mais ceux que lui procure le virus auxiliaire. C'est le seul exemple d'une atteinte proche des viroïdes en dehors de cellule végétale.
III - Liste des pathologies nerveuses liées aux prions :
Parmi les EST les plus connues, on peut citer :
III A - Chez l’humain :
1. La maladie de Creutzfeldt-Jakob
Décrite initialement en 1917-1918, c’est la maladie à prion humaine la plus fréquente (85 % des cas, si on ne prend pas en compte les maladies neurodégénératives du type Alzheimer, maladie de Parkinson, démence à corps de Lewy…) Elle présente plusieurs formes et sous-types qui se caractérisent initialement par des pertes de mémoire associées à une confusion (70 % des patients). 15 à 20 % présentent une incoordination des mouvements et une ataxie, qui se développe souvent au début de la maladie. Les myoclonies (= contraction musculaire brève et brusque) provoquées par le bruit ou d'autres stimuli sensoriels (sursauts myocloniques) apparaissent souvent au cours ou à des stades tardifs de la maladie. Il existe une démence précoce aboutissant au décès en quatre mois à deux ans selon la forme.
Plus d'infos en tableau : nombre de cas décédés certains ou probables de MCJ en France (mise à jour du 31 août 2021), Santé Publique France.
1a. sMJC : la Maladie de Creutzfeldt-Jakob sporadique :
C’est la plus commune (80 % des cas), atteignant le plus souvent le sujet âgé (plus de 40 ans mais médiane autour de 65 ans). Les troubles visuels : par exemple anomalies du champ visuel, diplopie, baisse de l'acuité ou flou visuel, agnosie visuelle (agnosie = trouble de reconnaissance avec incapacité à identifier un objet) sont fréquents dans cette forme. Des chercheurs (Douet, JY., Huor, A., Cassard, H. et al.) de l’INRAE et de l’ENVT (École Nationale Vétérinaire de Toulouse) ont identifié la charge infectieuse de différents tissus provenant de cinq patients décédés de sMCJ. Leurs résultats (3/2/2021) indiquent que le prion n’est pas confiné au niveau du système nerveux central, que de nombreux tissus périphériques comme les glandes salivaires, les reins, le cœur, le pancréas, la moelle osseuse peuvent être infectieux.
1b. La maladie de Creutzfeldt-Jakob familiale :
Elle survient dans environ 5 à 15 % des cas. L'âge de début est habituellement plus précoce que dans la maladie de Creutzfeldt-Jakob sporadique et la durée de la maladie est plus longue. La transmission héréditaire est autosomique dominante.
1c . La maladie de Creutzfeldt-Jakob acquise : 1 % des cas et deux formes « vMCJ » et « iMCJ » :
« vMCJ » (v pour variant) : en mars 1996, a été identifiée une autre forme de MCJ, un nouveau variant de la maladie de Creutzfeldt-Jakob. Il est nommé maintenant « vMCJ » et concerne le sujet jeune (moins de 30 ans). La transmission serait due probablement à l’ingestion de viande bovine contaminée par l’ESB (= encéphalopathie spongiforme bovine voir ci-dessous). La plupart des cas sont survenus au Royaume-Uni, qui comptait 178 cas au 3 février 2020 (deux depuis 2011), contre 54 cas dans tous les autres pays européens et non européens au 30 décembre 2019. Le temps entre l'ingestion de viande de bœuf contaminé et le développement de symptômes a été de 12 à 20 ans. La maladie dure environ un an et demi (anxiété, dépression plutôt que la perte de mémoire) et est mortelle.
« iMCJ » : La maladie de Creutzfeldt-Jakob iatrogénique (iMCJ) a été contractée par des greffes de cornée ou des transplantations de dure-mère (méninge la plus externe, blanc nacrée, très résistante, inextensible, qui enveloppe le cerveau) de cadavre + des électrodes stéréotaxiques intra-cérébrales + tout autre matériel contaminé + d'hormone de croissance préparée à partir de glandes hypophysaires humaines.
En juin 2019, une technicienne de laboratoire de 33 ans, Émilie Jaumain, était décédée des suites d’une maladie de Creutzfeldt-Jacob contractée en mai 2010. La jeune femme, alors en CDD, s’était entaillé le pouce en manipulant des cerveaux de souris infectées dans un laboratoire de l’Inrae à Jouy-en-Josas. Les neuf laboratoires français qui travaillent sur les maladies à prions, comme la maladie de Creutzfeldt-Jacob, ont suspendu depuis fin juillet 1921, leurs recherches pour trois mois. Un agent retraité d’un laboratoire de Toulouse de l’Inrae a déclaré la maladie, sans qu’il soit possible pour l’instant d’affirmer un lien entre son ancienne activité et ses symptômes.
2. L’insomnie fatale comporte deux types affectant dans le cerveau les « thalamus » :
Les deux thalamus (dénomination due à Galien) sont situés de part et d’autre du troisième ventricule (cavité où circule le liquide cérébrospinal) en constituant les parois, en dessous du noyau caudé et au-dessus de l'hypothalamus. Ses noyaux gris sont des relais pour les influx sensitifs et moteurs et interviennent dans la conscience, de la vigilance et du sommeil. Voir schéma IV C pour l’anatomie.
2a. L’insomnie fatale familiale (IFF) :
Encéphalopathie spongiforme transmissible, uniquement génétique, décrite en 1986, d’abord en Italie concerne une quarantaine de familles dans le monde dont plusieurs en France. Elle est à transmission autosomique dominante (un enfant de personne atteinte d'IFF a 50 % de risque d'en souffrir également) et liée, elle aussi, à une anomalie du gène de la protéine PrPC (gène PRNP) connue… une mutation ponctuelle des codons 178 et 179.
Elle se caractérisée par l'association d'une insomnie qui peut être initialement légère puis devient rebelle, évoluant vers une incapacité totale à trouver le sommeil + des phases anormales de sommeil paradoxal avec rêves et hallucinations durant l’éveil + des troubles végétatifs (disparition des rythmes circadiens, hyperactivité sympathique - par exemple HTA, tachycardie, hyperthermie, transpiration accrue = hyperhydrose - troubles sphinctériens) + des difficultés motrices (les myoclonies = spasmes musculaires saccadés sont rares, contrairement à la maladie de Creutzfeldt-Jakob) + un déclin cognitif et des symptômes psychiatriques qui peuvent être tardif. Les myoclonies sont rares. Le diagnostic peut être fait par des électro-encéphalogrammes (EEG).
Il existe une perte neuronale avec gliose des astrocytes en particulier dans le thalamus (noyaux dorso-médian et antérieur). La spongiose est discrète. Ici, c’est une particularité, il n’y a pas de plaques amyloïdes.
L'âge de début moyen est 51 ans (entre 18 et 60 ans). La maladie évolue vers une issue fatale par manque de sommeil en 6 à 32 mois. L'IFF a pu être transmise à des souris de laboratoire.
2b. L'insomnie fatale sporadique (SFI) :
Elle ne comporte pas de mutation génétique de PrP. L'âge moyen au début est légèrement plus avancé et l'espérance de vie est légèrement plus longue que dans l'insomnie fatale familiale. Les premiers symptômes comprennent un déclin cognitif et une ataxie. Les anomalies du sommeil ne sont généralement pas signalées, mais peuvent être observées au cours d'une étude polygraphique sur le sommeil.
3. Le syndrome de Gerstmann-Sträussler-Scheinker (SGSS) :
Décrit en 1936 et abordé en partie ci-dessus. Très rare forme (100 fois moins fréquente que la maladie de Creutzfeldt-Jakob). Le plus souvent familiale à transmission autosomique dominante. Cela signifie que si une personne est atteinte de cette maladie, les enfants ont au moins 50 % de chances de contracter la maladie dans leur vie. Elle survient à un âge plus précoce que la maladie de Creutzfeldt-Jakob (40 ans versus 60 ans), avec une espérance de vie plus longue (cinq ans versus six mois).
Elle se manifeste par une dysarthrie (= trouble de l'articulation de la parole correspondant à une atteinte nerveuse), un syndrome parkinsonien avec une ataxie (maladie neuromusculaire qui consiste en un manque de coordination fine des mouvements volontaires, elle aussi liée à une atteinte du système nerveux) et une hyporéflexie, puis par une démence. On observe un nystagmus, une paralysie du regard et une surdité. Les myoclonies sont beaucoup plus rares que dans la maladie de Creutzfeldt-Jakob. Elle affecte les patients de 30 à 60 ans. L'incidence exacte de la maladie est inconnue.
4. Le Kuru :
Décrit en 1957, entre autres par D. Carleton Gajdusek (prix Nobel de médecine 1976), il a aujourd’hui a disparu. Onze nouveaux cas de kuru ont été signalés entre 1996 et 2004, suggérant une période d'incubation qui peut dépasser 50 ans.
Il touchait les tribus Foré des hauts-plateaux de l’est de Nouvelle-Guinée (le mot kuru signifie « trembler de peur », en foré) qui avaient la particularité culturelle de manger le cerveau de morts lors de rites anthropophages mortuaires. Consommer des parents décédés visait de s’imprégner de leur force spirituelle et physique. La maladie touchait surtout les femmes et les enfants qui mangeaient le système nerveux central. Les hommes, consommant les muscles, étaient épargnés. Le nombre total de cas avoisine les 2 700 soit de l’ordre d’1/10 de la population. La maladie a disparu du fait des interventions des autorités provoquant l’arrêt des pratiques d’anthropophagie. Aucune anomalie diagnostique n'a été identifiée dans le gène PrP des personnes atteintes de kuru.
Symptômes : un syndrome cérébelleux débutant par des tremblements ressemblant à des frissons puis avec un trouble de l’équilibre, de la coordination des mouvements, des troubles visuels, des crises d'épilepsie et des secousses musculaires. Outre une marche instable, il fut signalé une grande instabilité émotionnelle et des rires incompréhensibles avec une mémoire qui s’affaiblit. Une démence peut compléter le tout, aboutissant au décès dans les deux ans après le début des symptômes. L'autopsie peut montrer des plaques typiques contenant PrPSc, avec une densité maximale dans le cervelet.
Il a été la première encéphalopathie spongiforme humaine dont la transmissibilité au singe a été démontrée : sept ans après avoir été inoculé par des extraits de cerveau d’un sujet atteint de kuru, un chimpanzé manifesta les troubles neurologiques du kuru.
5. « Maladie à prion associée à une diarrhée et à une neuropathie végétative » :
Une maladie à prion associée à une diarrhée et à une neuropathie du système autonome (= neuropathie végétative) a été identifiée en 2013 dans une famille britannique. Une maladie similaire a été rapportée dans une famille italienne.
Le prion amyloïde ne se limite pas au système nerveux central mais est distribué dans les nerfs périphériques et les organes internes. Les symptômes périphériques prédominent initialement et les symptômes au niveau du système nerveux central surviennent plus tard.
Cette maladie est associée à une nouvelle mutation dans le gène à prions (mutation Y163X pour la famille britannique : mutation Y162X dans la famille italienne), qui se traduit par une troncature de la protéine prion mutée. Ainsi, la protéine mutée est dépourvue de l'ancrage qui permet de se fixer aux membranes cellulaires, flotte dans les liquides corporels et migre vers d'autres tissus.
Les symptômes commencent à l'âge adulte ; ils comprennent la diarrhée chronique aqueuse, une rétention urinaire ou une incontinence urinaire, une hypotension orthostatique et une polynévrite périphérique principalement sensorielle. La détérioration des fonctions cognitives et les convulsions se produisent entre 40 et 50 ans.
6. La prionopathie à sensibilité variable à la protéolyse (VPSPr, variably protease-sensitive prionopathy) est une maladie à prions sporadique rare (1 à 2/100 millions de personnes), identifiée en 2008. La PrPSc est moins résistante à la digestion par les protéases, certaines variantes étant plus sensibles aux protéases que d'autres, d'où le nom : variably protease-sensitive. Elle ressemble à la maladie de Gerstmann-Sträussler-Scheinker (GSS) mais contrairement à cette dernière, aucune mutation du gène de la protéine prion n'a été identifiée.
Les patients présentent des déficits de la parole (aphasie et/ou dysarthrie), des troubles cognitifs et des symptômes psychiatriques. Une ataxie et un parkinson peuvent se développer. L'âge moyen de survenue est de 70 ans, et la durée de survie est de 24 mois. Environ de 40 % des patients ont des antécédents familiaux de démence.
7. D’autres maladies neurodégénératives liées à des prions ?
La capacité d’une protéine à changer de conformation, à s’agréger en recrutant la forme normale de la protéine et à se propager d’une cellule à l’autre n’est pas spécifique de la protéine prion. Les recherches ont montré que d’autres protéines de l’organisme impliquées dans des maladies neurodégénératives pouvaient adopter le même comportement : ainsi le peptide béta-amyloïde impliqué dans la maladie d’Alzheimer, l’alpha-synucléine dans la maladie de Parkinson. On dit qu’elles adoptent des comportements « prion-like ».
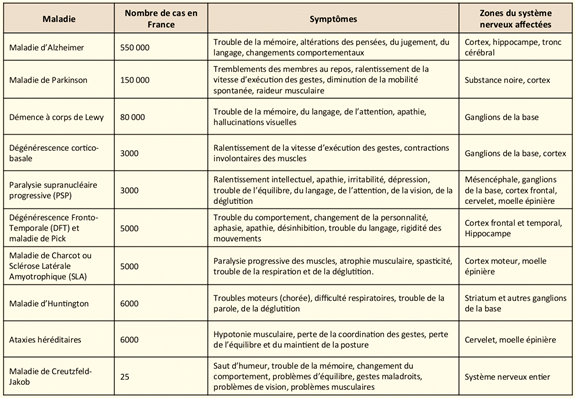
Les maladies neurodégénératives (MND) représentent un enjeu sociétal particulièrement important. Plus de deux millions d’aidants et 1,4 à 1,5 million de Français sont affectés par ces pathologies, et chaque année le nombre de nouveaux cas augmente à cause du vieillissement de la population, l’incidence étant directement liée à l’augmentation de l’âge.
Il s’agit d’un ensemble hétérogène d’affections de diverses populations de neurones. Lorsque, dans une MND, un groupe de neurones est sélectivement détruit, cela provoque des symptômes très divers comme la perte de mémoire, une paralysie musculaire, des troubles mentaux, etc.
Les représentants les plus connus des MND sont :
• La maladie d'Alzheimer
« Les maladies neuro-dégénératives » publié le 25.04.17 par Thibaut Burg
L’hippocampe est le substrat neuronal de la mémoire.• La maladie de Parkinson
• La démence à corps de Lewy
• La sclérose latérale amyotrophique ou maladie de Charcot
• La maladie de Huntington
• La maladie de Creutzfeldt-Jakob déjà citée
• Les démences fronto-temporales incluant la maladie de Pick (= démence frontotemporale à variante comportementale ou BvFTD) et d’autres variants. Dans la maladie de Pick, les patients deviennent impulsifs et perdent leurs inhibitions sociales… ils peuvent voler à l'étalage, frauder, négliger leur hygiène corporelle. Dans le syndrome de Klüver-Bucy ils présentent une labilité émotionnelle, une hyper-oralité avec succions et boulimie et une hypersexualité...)
 «
« Ces exemples font partie d’une liste de plus d’une centaine de maladies celles-ci étant souvent confondues. Il existe en effet une très mauvaise corrélation entre les symptômes observés par le clinicien et les affections neuronales retrouvées après analyses biologiques (EMG, EEG, IRM, analyses de biopsies ou prélèvements sanguins. Le diagnostic final ne pourra être validé définitivement que lors de l’examen post-mortem des patients.
Dans moins de 5 % des cas la maladie est due à une mutation dans un gène, qui peut être transmise à la descendance. Les 95 % restants sont des cas dit « sporadiques », les patients n’ayant pas d’antécédents génétiques familiaux, l’atteinte provient alors soit d’une mutation spontanée, soit de facteurs environnementaux ou d’autres facteurs encore inconnus. Pour une même maladie, des cas génétiques et des cas sporadiques peuvent se présenter.
 Liste non exhaustive de maladies neurodégénératives par Thibaut Burg (chiffres un peu sous-évalués car datant de 2014)
Liste non exhaustive de maladies neurodégénératives par Thibaut Burg (chiffres un peu sous-évalués car datant de 2014)J’avais fourni une partie des éléments suivants dans un article précédant concernant le "vaccin" contre la covid-19 de Pfizer et d’éventuelles conséquences du fait de la liaison avérée de la protéine de pointe du virus avec des protéines prion-like : « ARN vaccinaux de Pfizer producteurs de protéines spikes ».
1. À propos de la maladie d’Alzheimer :
Dans la maladie d’Alzheimer (MA), dans plus de 90 % des cas, la maladie débute par des troubles de la mémoire des évènements récents, on parle de « mémoire épisodique ». Par contre les souvenirs anciens ou les connaissances générales sont préservés beaucoup plus longtemps.
Avant même l’apparition des premiers symptômes, les neurones sont affectés depuis plusieurs années par deux types de lésions concomitantes, en fait deux types de prions : les plaques amyloïdes = plaques séniles (oligomères solubles de peptides β-amyloïdes) que l’on retrouve entre les neurones dans la substance grise du cortex cérébral + la « dégénérescence neurofibrillaire ou DNF » (la protéine à l’origine de ce dysfonctionnement est appelée « protéine Tau » que l’on retrouve à l’intérieur des neurones. Ces deux lésions correspondent à des amas de protéines qui se forment lors du processus normal du vieillissement. Cependant, dans les maladies de type Alzheimer, ces protéines s’accumulent en beaucoup plus grande quantité.
Dans onze échantillons de malades d’une autre maladie (angiopathie cérébrale amyloïde) seuls des prions bêta-amyloïdes ont été détectés. Chez des malades ayant souffert d’une dégénérescence fronto-temporale, seuls les prions Tau sont apparus. “Je pense que cela montre, sans l’ombre d’un doute, que les protéines bêta-amyloïdes et Tau sont toutes les deux des prions et qu’Alzheimer est une maladie à double prions dans laquelle ces deux mauvaises protéines détruisent ensemble le cerveau” Stanley Prusiner, 2019.
La protéine Tau joue normalement un rôle dans la stabilisation des microtubules permettant la croissance et le transport dans l’axone des neurones. Son action est régulée par des mécanismes de phosphorylation. Elle peut s’accumuler sous forme anormale, hyperphosphorylée et s’agréger en provoquant la mort neuronale.
La protéine Tau est incriminée dans de nombreuses démences, dont la maladie Alzheimer et la maladie de Pick (forme de démence frontotemporale). Dans cette dernière maladie, des inclusions constituées d'une forme anormale de protéine Tau s’accumulent dans le lobe frontal et les lobes temporaux. Ces inclusions sphériques et argentées, appelées « corps de Pick », entraînent le gonflement des neurones (cellules de Pick) puis leur dégénérescence.
La protéine Tau normale, par le biais de l’insuline, joue un rôle dans la plasticité cérébrale.
Il est maintenant possible de détecter très tôt la maladie d’Alzheimer, avant même la survenue des signes cliniques grâce à trois marqueurs. Ce dosage s’effectue dans le liquide céphalorachidien = LCR (ponction lombaire, prélèvement dans un tube de polypropylène) et concerne : les protéines Tau totales (= t Tau) + les protéines Tau hyperphosphorylées = pTau (qui toutes deux augmentent de façon modérée) + le peptide Aß 1-42 (qui séquestré en plaques amyloïdes baisse beaucoup). Ces biomarqueurs sont aussi utilisés pour distinguer la MA d’autres maladies neurodégénératives comme la maladie de Creutzfeldt-Jakob (MCJ), la démence à corps de Lewy et la démence vasculaire. Ainsi, dans la démence à corps de Lewy, Tau est normale ou augmentée, tau phosphorylée est normale et Aß1-42 baisse modérément. Dans le cas d’un accident vasculaire cérébral, Tau augmente beaucoup alors que les deux autres marqueurs sont normaux.
Protéine bêta-amyloïde sous forme de prions (en jaune).
 Protéine bêta-amyloïde sous forme de prions (en jaune).
Protéine bêta-amyloïde sous forme de prions (en jaune).Plus d'infos sur le sujet :
[25] Juebin Huang , MD, PhD, Department of Neurology, University of Mississippi Medical Center. Dernière révision totale mars 2021.
https://www.msdmanuals.com/fr/professional/troubles-neurologiques/syndrome-confusionnel-et-d%C3%A9mence/d%C3%A9mence-frontotemporale
[26] https://www.sciencedirect.com/science/article/abs/pii/S0035378716000217 Avril 2016 par Lou Grangeon, Claire Paquet, Stéphanie Bombois et David Maltete.
« Un quart des cas diagnostiqués « Alzheimer » seraient en fait dus à une nouvelle maladie, baptisée LATE (encéphalopathie à prédominance limbique). En cause, TDP-43 (Transactive Response DNA-binding Protein 43), une protéine indispensable à l'activité de nos cellules, qui se révèle aussi une vraie tueuse de neurones sous une forme anormalement phosphorylée. Nous y reviendrons.
La sclérose latérale amyotrophique ou SLA est une maladie des motoneurones hautement débilitante. La caractéristique pathologique de la SLA est la présence d'inclusions dans le cytoplasme des neurones moteurs spinaux survivants. Là encore, TDP-43 (encore nommée TAR DNA-binding protein-43) est la protéine majeure trouvée dans ces inclusions cérébrales post-mortem de patients atteints de SLA. Les RRM sont deux motifs de reconnaissance à l’ARN portés par TDP-43. L'altération pathologique (repliement anormal puis agrégation) de TDP-43 est considérée comme une caractéristique des maladies neurodégénératives de la sclérose latérale amyotrophique (et de la dégénérescence lobaire fronto-temporale). Nous détaillerons au chapitre V D.
2. À propos de la maladie de Parkinson et de la démence à corps de Lewy (DCL) :
En pathologie humaine, on sait que les protéines d’alpha-synucléine mal repliées sont converties en oligomères pathologiques puis en agrégats de fibrilles qui s'accumulent à leur tour dans les corps de Lewy (Lee VM, Trojanowski JQ. ; 2006). Ces dépôts interrompent les messages transmis par les neurones. Les corps de Lewy sont donc des inclusions neuronales, habituellement sphériques, des structures amyloïdes, principalement constitués de filaments neuronaux + la protéine prion-like appelée alpha-synucléine, protéine présynaptique qui aurait en conditions normales un rôle physiologique dans l’apprentissage et biochimique dans la régulation des niveaux de dopamine.

Chez les patients atteints de la maladie de Parkinson (prévalence supérieure à 2 % après 65 ans), les corps de Lewy se trouvent dans les cellules localisées dans le tronc cérébral (à la base du cerveau) au niveau de la « substance noire » = substantia nigra = locus niger, qui joue un rôle dans le contrôle des mouvements. À ce niveau, une petite population de neurones est affectée, entraînant la diminution d’un neurotransmetteur, la dopamine. Le traitement des patients avec un précurseur de la dopamine, la lévodopa, améliore considérablement certains des symptômes de la maladie. Le bénéfice du traitement se réduit lorsque la neurodégénérescence progresse. « Des thérapies d’appoint innovantes et peu couteuses pourront améliorer l’état des patients parkinsoniens en utilisant des stimulations répétées du cervelet ».
Dans la maladie à corps de Lewy, aussi appelée « démence à corps de Lewy (DCL) » une démence cliniquement proche de la maladie d’Alzheimer, les corps de Lewy sont présents dans la couche externe du cerveau appelée cortex responsable des grandes fonctions mentales.
III B - Chez l’animal :
1. La tremblante (gratte ou scrapie) du mouton, de la chèvre :
Diagnostiquée pour la première fois en Grande-Bretagne en 1732. Cuillé et Chelle ont injecté, par inoculation intraoculaire, à des moutons, des homogénats (tissus broyés) de cerveaux de moutons morts de la tremblante. Les animaux inoculés ont à leur tour déclaré la maladie démontrant ainsi que la tremblante était due à un agent infectieux.
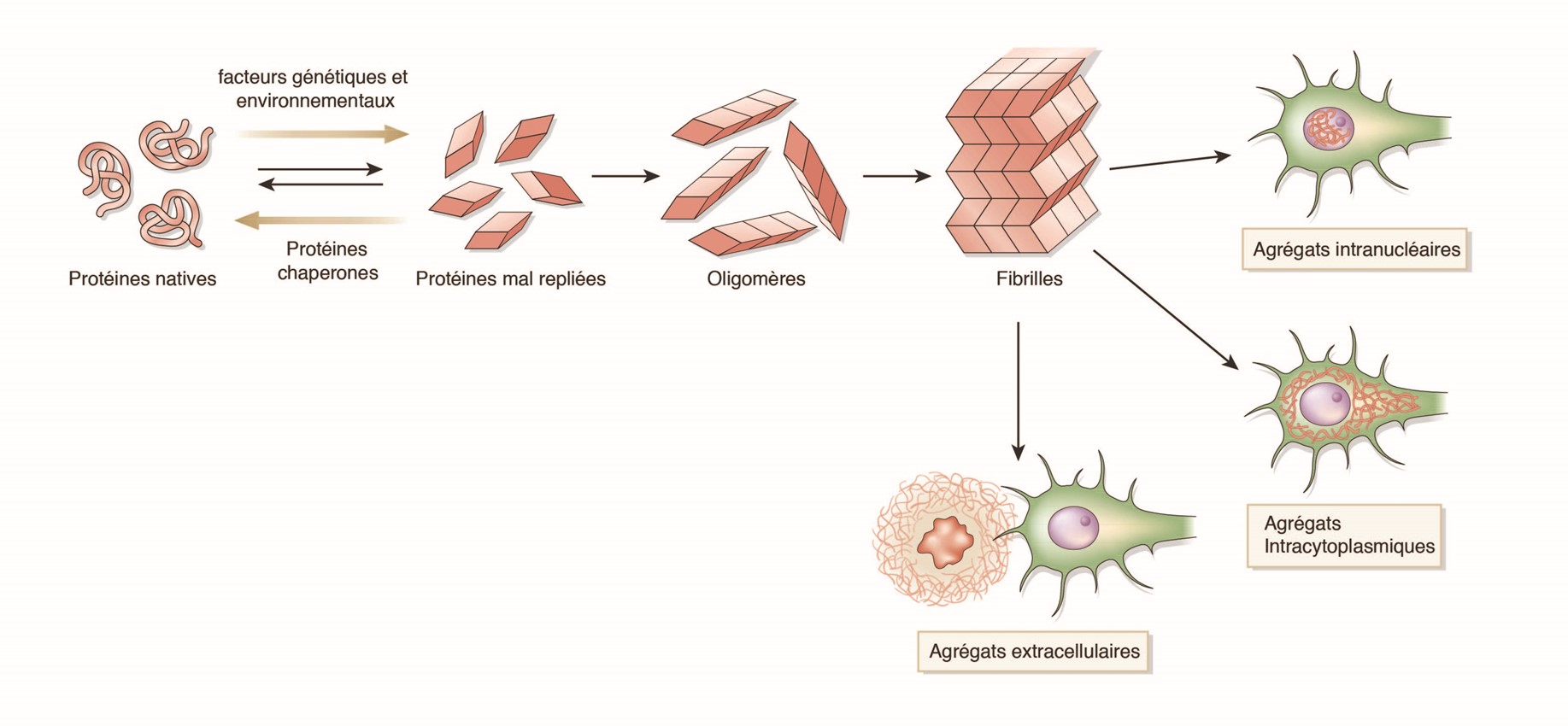 Mécanismes de formation des agrégats de protéines dans les maladies neurodégénératives
Mécanismes de formation des agrégats de protéines dans les maladies neurodégénérativesMalgré l’hétérogénéité de ces maladies neurodégénératives, il est possible de repérer une physiopathologie commune : une agrégation toxique de protéines qui induit la mort des neurones par apoptose. Dans des conditions physiologiques, les protéines mal repliées sont soit corrigées par des protéines chaperonnes soit dégradées et éliminées par le système ubiquitine-protéasome ou par le système phagosome-lysosome (autophagie). Les protéines qui doivent être dégradées sont marquées par une protéine, l’ubiquitine, qui permet de les diriger vers le protéasome. Ce dernier est un complexe enzymatique qui va permettre de couper les protéines en peptides de 7 à 9 acides aminés qui seront ensuite hydrolysés puis recyclés hors du protéasome. « Des facteurs tels que l’âge, les mutations génétiques, des changements de l’environnement intracellulaire, les variations de pH, le stress oxydant, ou les métaux lourds, peuvent conduire à l’augmentation de concentration de protéines mal repliées. » Ces protéines normalement solubles vont alors s’agréger entre elles pour constituer des oligomères qui vont eux-mêmes s’agréger formant ainsi des fibrilles qui sont insolubles. Ces dernières vont alors former des inclusions intranucléaires ou intracytoplasmiques et peuvent également constituer des agrégats extracellulaires qu’on appelle alors plaques séniles.
Schéma de T. Burg 25.04.17.
La transmission de la tremblante s'effectue essentiellement de la mère au jeune par la voie placentaire, mais aussi via le lait chez les ovins et les caprins.
Le prion est également présent dans le sang et l’urine. Le temps d'incubation est de 1,5 an (en moyenne). Le plus jeune sujet à avoir été diagnostiqué était un agneau âgé de sept mois. Aucun cas de transmission à l'Homme n'a été signalé depuis plus de deux siècles. Mais l'identification de l'agent infectieux (le prion) est très récente. C’est en 1967 que le mathématicien Griffith émit pour la première fois l’hypothèse que cet agent pourrait être uniquement constitué d’une protéine. Il fallut 40 ans pour que Stanley Prusiner le prouve. On n'a jusqu'à présent jamais démontré la possibilité d'une transmission de la tremblante à l'Homme ou à d'autres animaux (sauf souris), mais elle ne peut être exclue avec certitude.
Dans les deux formes (voir suite), on observe, dès les premiers symptômes, des troubles du comportement (fuite, désorientation, reculs) ainsi qu'une hyperesthésie se manifestant par des tremblements provoqués par la moindre excitation (oreilles, puis tête, encolure et membres), d'où le nom français de la maladie : tremblante.
On distingue habituellement deux formes cliniques de la tremblante : la forme prurigineuse et la forme paralytique, mais il est possible d'observer l'expression des deux formes chez un même animal. Dans la forme prurigineuse, la maladie débute par prurit dorso-lombaire, qui tend à se généraliser, avec développement de comportements de grattage (d'où le nom anglo-saxon de la maladie, scrapie, du verbe to scrape : gratter). La laine est ébouriffée, puis arrachée par plaques, laissant la place à des lésions infectées. La forme paralytique débute, quant à elle, par une parésie des membres postérieurs (démarche ébrieuse, chutes, perte de la coordination).
L'appétit est conservé, mais l'état général de l'animal s'altère et les symptômes s'aggravent : tremblements permanents, décubitus, amaigrissement, cachexie, coma et convulsions, le décours, toujours fatal, en 15 jours à 6 mois.
Les souris sont sensibles à la tremblante du mouton, leur gène Sinc contrôlant la période d'incubation de la maladie a été identifié en 1968. En 1982, il a été montré que Sinc code pour la protéine PrP, qui copurifie avec la fraction prion.
Les animaux dépourvus du gène codant pour la PrP ne sont généralement pas sensibles aux agents EST, ont une durée de vie normale, ne présentent pas d'anomalies dans le système neural et ne propagent pas le prion. En revanche, lorsque des animaux qui codent pour la PrP de type sauvage sont infectés par l'agent EST, ils accumulent des agrégats de PrP résistants aux protéases dans les tissus neuronaux. Ces agrégats sont constitués de la forme prion de la PrP, désignée PrP Sc.
Plus d'infos : https://journals.asm.org/doi/10.1128/msphere.00520-20?permanently=true
2. L'encéphalopathie spongiforme bovine (ESB) = Bovine spongiform encephalopathy ou BSE en anglais, communément connue sous le nom de « maladie de la vache folle » :
Des centaines de milliers de bovins ont développé l'encéphalopathie spongiforme bovine. La seule façon de confirmer la présence de l'ESB est d'examiner le tissu cérébral de l'animal après sa mort. Les scientifiques pensent que l'ESB est surtout propagée en donnant à manger aux bovins différentes farines de viandes dérivées d'animaux abattus (parties non consommées des carcasses bovines + cadavres d'animaux comme les moutons, les chèvres et d'autres bovins). Au cours de ce processus, une protéine anormale liée à l'ESB peut être transmise d'un animal malade abattu à un animal sain.
Les symptômes extérieurs apparaissent généralement quatre à cinq ans après la contamination, et toujours sur des animaux de plus de deux ans (entre trois et sept ans généralement). Comme l'ESB provoque des lésions au tissu cérébral, il cause une variété de symptômes allant de phénomènes comportementaux à des problèmes de coordination. Ils se manifestent au début par une modification du comportement de l'animal avec de la nervosité ou un comportement agressif… il peut parfois donner des coups de pied, manifester une appréhension et une hypersensibilité aux stimulations externes (bruit, toucher, éblouissement) et s'isoler du reste du troupeau. Il a des difficultés de coordination ou pour garder la position debout, une diminution de la production de lait et une perte de poids.
La maladie est mortelle et la mort se produit deux semaines à six mois après l'apparition des symptômes.
Les scientifiques se sont aperçus début 1996 de la possibilité de transmission de la maladie à l'Homme par le biais de la consommation de produits carnés. La maladie a fait 231 victimes humaines en 2016.
Voir aussi : Question écrite n° 30973 de M. Fernand Demilly (Somme - RDSE) et réponse du ministère de la Santé publiée dans le JO Sénat en 2001 : « Aucun des vaccins actuellement commercialisés en France n'utilise des produits bovins d'origine britannique ».
3. L’encéphalopathie spongiforme féline :
L'ESF chez un chat a été diagnostiquée pour la première fois en 1990 en Grande-Bretagne. Ce pays a enregistré, à ce jour, quelque 90 autres cas chez des chats domestiques. Un cas a été découvert en 1995 en Norvège et un autre en 1996 chez un chat mâle dans la principauté de Liechtenstein. Des cas d'ESF ont été aussi diagnostiqués dans des zoos chez des félins sauvages (puma, ocelot, guépard, lion, tigre) qui avaient été nourris avec des déchets d'abattoirs crus. Selon les connaissances actuelles, l'agent responsable de l'ESF a une grande affinité avec celui de l'ESB. L’incubation moyenne est de cinq ans. Chez les chiens, ou les canidés en général, ce tableau clinique n'a jamais été observé.
4. L’encéphalopathie spongiforme du vison : contamination se faisant à la suite de la consommation par les visons de viande de moutons ou de bovins infectés. Le premier foyer est apparu aux États-Unis en 1947. La période d'incubation est de sept à douze mois.
5. Le dépérissement chronique du cervidé (CWD pour Chronic Wasting Disease) = maladie débilitante chronique des cervidés = "cachexie chronique" qui affecte la faune sauvage en liberté et circule dans l'environnement en Amérique du Nord et en Europe du Nord. Des hamsters alimentés avec des plantes croissant sur un sol dans lequel était enterré un cerf mort de CWD (équivalent de la vache folle chez le cerf) sont tombés malades, ce qui évoque une migration du prion du sol vers la plante. Contamination même si les plantes sont lavées cinq fois complètement et séchées avant d'être données comme aliment aux hamsters… Les plantes pourraient donc jouer un rôle dans la transmission horizontale de la maladie.
Suite de l'article : Que sont les prions ? Partie II.
NDLR : modifications apportées le 29.09.21 à 10h00.
À LIRE AUSSI


L'article vous a plu ? Il a mobilisé notre rédaction qui ne vit que de vos dons.
L'information a un coût, d'autant plus que la concurrence des rédactions subventionnées impose un surcroît de rigueur et de professionnalisme.
Avec votre soutien, France-Soir continuera à proposer ses articles gratuitement car nous pensons que tout le monde doit avoir accès à une information libre et indépendante pour se forger sa propre opinion.
Vous êtes la condition sine qua non à notre existence, soutenez-nous pour que France-Soir demeure le média français qui fait s’exprimer les plus légitimes.
Si vous le pouvez, soutenez-nous mensuellement, à partir de seulement 1€. Votre impact en faveur d’une presse libre n’en sera que plus fort. Merci.

Tous les vendredis, recevez la newsletter hebdo de France-Soir
