Histoire du COVID-19 - chapitre 9 - Partie 1 : Un tropisme du SARS-Cov2 vers certaines caractéristiques infectiologiques du VIH1

Où le mathématicien Jean-Claude Perez et le professeur Luc Montagnier nous révèlent que les laboratoires de virologie font planer un danger obscur sur l'humanité.
Résumé du lecteur pressé - Nous avons consacré les parties 2, 3 et 4 du chapitre 8 à démontrer que sur le plan scientifique le SARS-Cov2 ne pouvait être qu'un coronavirus manipulé par génie génétique. Nos conclusions obtenues à partir d'une analyse et de calculs indépendants sont corroborées par celles du Dr Steven Quay. Il vient de publier un article de 193 pages où il calcule, par une méthode différente de la notre, que la probabilité que le SARS-Cov2 provienne d'un laboratoire est de 99,8%. Les observations anormales s'accumulent inexorablement comme le fait improbable que l'Orf1a du génome du SARS-Cov2 contient une séquence de 71 nucléotides 100% identique à une section du rétrovirus endogène humain HERV... La réalité des calculs est exposée au vu de tous mais cela ne suffit pas pour l'establishment scientifique international qui craint de devoir demander l'audit de l'Institut de virologie de Wuhan. Nous étudions dans ce chapitre une autre face de la réalité, plus difficile à cerner, qui suggère qu'un danger plane sur une humanité dépourvue de défense devant des virus synthétiques nouveaux qui, plus que des chimères, seront des cocktails transgéniques potentiellement capables de s'intégrer au génome humain. La présence de mini-séquences concentrées du VIH1 et du VIH2 dans le SARS-Cov2, n'est peut-être pas seulement un tropisme naturel des coronavirus à SARS qui résulterait d'une parenté avec le VIH dans l'évolution. Là encore, pourrait-il y avoir un gain-de-fonction méthodiquement organisé ?
Les premiers à avoir suggérer cette possibilité sont Pradhan et al. dans un article déposé sur le site BioRxiv avant qu'ils ne soient obligés de se rétracter. L'article de Prashant Pradhan et al., bien que très critiquable par certains aspects montre la réalité de la présence de ces séquences d'acides aminés du VIH à des endroits clé de la protéine S.
Les seconds sont le mathématicien Jean-Claude Perez et le professeur et prix Nobel Luc Montagnier qui attirent l'attention sur la concentration anormale de mini-séquences de nucléotides apparentées au VIH dans le gène de la protéine S, en évoquant la possibilité que leur présence soit en relation avec une tentative d'élaboration d'un vaccin anti-sida. Jean-Claude Perez et le professeur Luc Montagnier vont plus loin dans l'analyse du génome du SARS-Cov2 avec des implications qui nécessitent l'éclairage d'un savant généticien.
Présence de mini-séquences du virus VIH présentes dans le génome du SARS-Cov2 - une stratégie vaccinale possible ?
Dans son interview télévisée du 17 avril 2020, le Pr Luc Montagnier a attiré l'attention sur la présence effective d'au moins une demi-douzaine de mini-séquences du virus du VIH regroupées dans un segment très court du génome du SARS-Cov2. Cette observation avait été publiée par le mathématicien Jean-Claude Perez en février 2020 sous le titre « Origine synthétique du Covid-19 et Évolution ». Notons que la présence de ces très petites séquences apparentées au VIH1 et au VIH2, bien qu'étant un fait qui soulève des questions, n'est pas le point central de cette étude que nous expliquerons dans la prochaine partie de ce chapitre et qui révèle un aspect remarquable de l'évolution des espèces.
A proprement parler, il ne s'agit pas d'insertions comme on a pu le dire mais de petits fragments de séquences similaires ou identiques au VIH, ce qui est différent. Selon Luc Montagnier et Jean-Claude Pérez ces mini-séquences d'une longueur comprise entre 18 et 30 nt se trouvent statistiquement au-dessus du seuil de détection d'un signal génétique réel. Elles pourraient être des éléments informatifs exogènes (EIE), c'est-à-dire qu'ils peuvent avoir une signification génétique et donc une influence physiologique. Ils affirment que cette présence indubitable d'EIE concentrés, en relation avec le VIH mais également avec le parasite Yoeli Plasmodium (1 longue mini-séquence de 38 nt), l'agent responsable du paludisme, ne serait pas naturelle et traduirait une stratégie de tentative d'élaboration de vaccin qui aurait fuité d'un laboratoire de recherche en Chine.
Principe général de la vaccination – exemple du Mosquirix, le vaccin anti-paludique raté.
En effet, le principe vaccinal repose essentiellement sur la génération d'anticorps neutralisant qui reconnaissent de très courtes séquences d'acides aminés (aa), typiquement 4 à 12 aa (équivalent à une longueur de 12 à 36 nt), formant des épitopes antigéniques qui peuvent être introduits dans une protéine exposée (protéine d'enveloppe) appartenant à un autre virus par ailleurs inactivé. Le développement d'un vaccin anti-VIH prophylactique nécessiterait la mise au point d'épitopes antigéniques capables de générer des anticorps à large spectre, c'est à dire peu sensibles à la variation de leur composition en acides aminés et pouvant attaquer le virus en de multiples endroits. Ce problème est si délicat qu'il fait l'objet d'une recherche de pointe (thèse doctorale de Fahd Benjelloun 2013).
« Le développement d’un vaccin prophylactique efficace, réside aussi dans le choix d’un immunogène capable de mimer sur le plan conformationnel des épitopes majeurs du VIH1. » et la sélection d’anticorps monoclonaux neutralisants à large spectre (Thèse de Fahd Benjelloun 2013).
L'affirmation de Luc Montagnier ne vient pas au hasard parce qu'en réalité, comme nous l'avons déjà mentionné précédemment (chapitre 7 partie 2), le virus du sida cause des ravages en Chine qui ne sont pas communiqués à l'OMS. Un autre fléau montant en Chine est le paludisme en provenance d'Asie du Sud-Est et d'Afrique avec laquelle la Chine a développé de très nombreux contacts économiques et stratégiques ces 15 dernières années. L'épidémie de paludisme en Chine semble avoir été jugulée depuis 2015, pour un coût de 135 millions de dollars (USD). Mais la possibilité de résurgence existe forcément et la nécessité de développer un vaccin subsiste vu le coût économique et humain de cette maladie. Notons que les pays d'Afrique et la Chine partagent le même double combat au sujet du sida et du paludisme qui sont leurs fléaux communs. De ce fait, on peut se demander s'il existe un accord tacite entre l'OMS, dont le directeur Tedros Ghebreyesus est Ethiopien, et le gouvernement chinois pour l'élaboration de vaccins contre le sida et le paludisme tant ses déclarations et ses actes semblent inféodés au pouvoir chinois. Comme nous avons pu le constater avec le SARS-Cov2 les fléaux sanitaires ont des répercussions économiques et sociales gravissimes au niveau mondial et constituent donc un enjeu géopolitique.
Autre point commun, bien que les agents de ces deux maladies différent, l'un est un virus et l'autre un parasite, ils résistent depuis des décennies à l'élaboration de vaccin en raison de leurs mécanismes particuliers de réplication et de diffusion dans l'organisme.
Le virus du sida est un casse-tête car il est extraordinairement variable au niveau de sa glycoprotéine de surface gp120 qui lui permet de se fixer au récepteur CD4 pour infester les lymphocytes T4.Les parties les plus exposées au cytoplasme (les boucles) de cette protéine mute énormément. Ce pouvoir de mutation viable du virus est tel qu'il est mesurable au cours du développement de l'infection au sein d'un même individu. Les mécanismes d'évasion du virus du système immunitaire sont multiples et permettent d'échapper à l'immunisation d'anticorps générés au moment de la primo infection. Non seulement cela empêche la production d'anticorps d'efficacité durable mais, de plus, le virus VIH pénètre dans les cellules qui sont les médiatrices de la réponse immunitaire acquise. C'est-à-dire qu'elles sont chargées de déclencher la réponse qui sera faite à partir à partir des anticorps engendrés par la détection d'antigènes via les complexes majeurs d'histocompatibilité (MHC). Les cellules T CD4+ (i.e. porteuse du récepteur CD4) sont chargées de détecter les protéines de surface des virions pour diriger vers eux d'autres cellules immunitaires comme les lymphocytes B pour les détruire. Comme elles ne peuvent pas détecter un virus caché en leur sein, elles sont inopérantes et le VIH les détruit en s'y répliquant massivement à l'intérieur, conduisant à l'arrêt totale du système immunitaire acquis qui engendre le syndrome d'immunodéficience.
Dans le cas du paludisme, la difficulté de mise au point d'un vaccin est due au fait que le parasite migre d'un organe à un autre en exprimant différents antigènes aux différents stades de l'infection. Il pénètre les globules rouges ce qui lui permet d'échapper au système immunitaire dans le sang. Le cycle du parasite est différent selon les organismes qu'il infecte de sorte que la recherche ne dispose pas de modèle animal très fiable, les rongeurs n'étant pas sensibles aux mêmes souches que l'homme. Comme pour le virus du sida une stratégie vaccinale serait de déclencher une réaction immunitaire basée sur un grand nombre d'antigènes différents pour tenter de piéger l'agent pathogène massivement dès le début de l'infection avant qu'il ne se transforme. Mais c'est un mirage par ce qu'il suffit qu'une proportion infime du virus ou de l'agent pathogène échappe au premier tire de barrage de l'organisme pour rendre la vaccination caduque. Ainsi, le développement d'un vaccin contre le paludisme bien qu'à l'étude depuis 60 ans n'a jamais pu démontrer autre chose que l'ego démesuré de gens qui y ont consacré leur vie. Ils ne veulent pas s'avouer vaincu, voulant démontrer à toute force la supériorité de l'homme sur la nature.
Le vaccin antipaludique Mosquirix, testé à nouveau en Afrique en 2019, est fabriqué par GSK et sponsorisé par la fondation Bill Gate. Il est élaboré sur le principe « de combiner des séquences antigéniques extraites de protéines du parasite, qui engendre une réponse immunitaire par anticorps, avec l’antigène de surface du virus de l’hépatite B pour former un virus pseudo typé vaccinal – une combinaison [NDA, un mélange transgène] qui, une fois injectée est supposée déclencher une réponse cellulaire du système immunitaire, et générer des anticorps. »(Source : Le Blob)
Cette stratégie vaccinale pourrait donc être celle à laquelle se réfère le professeur Montagnier en ce qui concerne la présence d'EIE du HIV dans le SARS-Cov2. Elle est en tout cas digne d'une recette d'apprenti sorcier car, quoiqu'on en dise, l'utilisation de virus dangereux (HIV, hépatite B, …) pseudotypés, dont on se demande toujours jusqu'à quel point ils sont inactivés, pose des problèmes de morale. En tout cas, ce vaccin fruit de décennies d'efforts « même s’il permet d’éviter des millions de contaminations n’offre qu’une protection partielle qui décline avec le temps. Les résultats font également état d’un nombre accru de cas de méningites et de paludismes cérébraux parmi les personnes ayant reçu le traitement ». (Source : Le Blob)
Les chercheurs de GSK avancent l’idée d’une quatrième dose, administrée 18 mois après la troisième, « pour stabiliser les défenses immunitaires.» (Source : Le Blob) Il y a comme une contradiction car l'évitement des contaminations ne dure que le temps de la protection partielle et entraîne la nécessité de revaccination à intervalle régulier avec tout ce que ça implique en matière de danger auto-immun dû à la répétition annuelle abusive de vaccination, sans parler des autres dangers que nous avons développés dans la partie 1 de ce chapitre. Cette répétition abusive de vaccination est exactement ce qui semble se profiler avec le vaccin contre le SARS-Cov2, cela se fera avec la complicité des gouvernements pour le plus grand profit de Big Pharma.
Les chercheurs indiens Pradhan et al. obligés de rétracter leur article sur les insertions de séquences du VIH1 dans le SARS-Cov2.
La comparaison des virus doit impliquer la comparaison des séquences protéiques. Avant d'examiner plus en détail les EIE repérés dans les travaux conjoints de Montagnier et Perez, rappelons que l'on peut comparer la proximité des organismes au niveau de leurs génomes mais aussi au niveau des protéines que les gènes expriment. Elles constituent les éléments fonctionnels du vivant en structurant les cellules, assurant leur communication, leur développement et qui interviennent dans une multitude de réactions chimiques physiologiques. La comparaison au niveau protéique est essentielle pour comprendre et mesurer l'impact des changements au niveau génomique (mutations ponctuelles, insertions et délétions de groupes de nucléotides) sur la fonction et les caractéristiques fonctionnelles du gène exprimé.
Dans l'analyse d'un virus les deux types de comparaisons doivent être menés en parallèle. Par exemple, comme nous l'avons vu en partie 3, les protéines S du SARS-Cov2 et du RaTG13 partageant 97.7% d'identité en acides aminés (99.1% en dehors du RBM et du site de la furine) sont en réalité bien plus proches l'une de l'autre que ce que la comparaison au niveau génomique ne le laisse supposer (93.2%). Ainsi, on se rend compte à quel point le SARS-Cov2 est dramatiquement relié au RaTG13 avec plus de 99% d'identité également sur les autres protéines structurales qui sont E (100%), M (99,1%) et N (99,4%) ainsi que sa polymérase (99,6%). Avec la protéine S ces protéines garantissent l'essentiel du fonctionnement du virus. Cela est d'autant plus troublant qu'au moins 7 années d'évolution sont censées séparer ces deux virus. Cette proximité extraordinaire entre un virus humain et un virus de chauve-souris stocké dans un laboratoire de recherche fait fi de la pression de sélection positive qui aurait dû caractériser la protéine S en franchissant la barrière d'espèce. Probablement, aucune théorie argumentée ne pourra jamais expliquer raisonnablement comme une telle proximité peut apparaître naturellement entre deux virus qui par ailleurs ne devrait pas avoir de rapport direct.
C'est également par la comparaison au niveau de la séquence protéique de la protéine S que le chercheur indien Prashant Pradhan et son équipe sont arrivés à identifier la présence « étrange » de 4 mini-séquences identiques ou similaires présentes dans les protéines gp120 (glycoprotéine de reconnaissance du récepteur CD4) et gag (protéine de fusion à la membrane cellulaire) du VIH1. Ils ont été les premiers avec Lyons Weiler à tenter de publier des observations critiques sur le SARS-Cov2. Leur article pre-publié (preprint) a été déposé le 31 janvier 2020 sur le site BioRxiv, soit tout juste 2 semaines après le dépôt de la séquence du SARS-Cov2 dans une banque de donnée internationale (GenBank). Ils ont été obligés de le retirer sous la pression politique et scientifique en raison du déchaînement de peur et de spéculation complotistes que leur conclusion pouvait engendrer.
Pour ce qui est de leurs observations, elles correspondent à la première étape d'une analyse fonctionnelle sur la pathogénie du SARS-Cov2 en comparant sa protéine S avec celle du virus le plus proche qui selon eux était le virus GZ02. Le virus GZ02 est un variant du virus SARS-Cov isolé à Guangzhou en septembre 2003. Comme le SARS-Cov Urbani, considéré comme le génome de référence pour l'épidémie de 2002-2003, la protéine S de ce virus partage 75% d'identité de séquence aa avec celle du SARS-Cov2. Pourtant au niveau du sous domaine S1-N-ter jusqu'au RBM (région de Lyons-Weiler) la situation n'est pas du tout la même avec 61% d'identité pour le GZ02 contre 75% pour le SARS-Cov (Urbani). Donc, de ce point de vue, cela prouve qu'ils ont tenté de rendre, comme nous allons l'expliquer, leur démonstration plus probante.
Ils savaient parfaitement, grâce aux logiciels d'alignements multiples, que le sous-domaine S1-N-ter du SARS-Cov était celui le plus proche du SARS-Cov2. Cela semble indiquer qu'ils connaissaient à l'avance ce qu'ils cherchaient, c'est-à-dire des mini-séquences du VIH insérées à des endroits clés comme le sous-domaine S1-N-ter. N'oublions pas qu'il s'est écoulé à peine 15 jours entre le dépôt de la séquence du SARS-Cov2 à GenBank et le dépôt de leur article sur BioRxiv... Il s'agit là d'un travail de recherche hypersonique, peu compatible avec une réelle recherche. Cela, indiquerait que les insertions de mini-séquences du VIH sont bien une technique vaccinale expérimentale envisagée dans le milieu confidentiel de la recherche en virologie. Cette technique rejoint très logiquement celle que nous avons exposée précédemment pour le vaccin contre le paludisme.
Pradhan et al. montrent que par rapport au GZ02 la protéine S du SARS-Cov2 contient cinq insertions de longueur comprise en 3 et 7 acides aminés (résidus). Les insertions dans le code ARN d'un virus peuvent se produire tout à fait naturellement au cours de la réplication de l'ARN des virus. Quatre insertions, parmi les cinq insertions qui augmentent la longueur de la protéine S du SARS-Cov2 par rapport au SARS-Cov, correspondent à des séquences de gp120 et gag et sont d'une longueur comprises entre 6 et 12 aa et, qui plus est, sont uniques au SARS-Cov2 parmi tous les autres coronavirus, selon Pradhan et al. Nous avons procédé à la vérification de ces affirmations qui sont exactes.

Par contre, deux des 4 insertions bien qu'étant unique au SARS-Cov2 parmi tous les coronavirus ne sont pas uniques au VIH1 car on les retrouve dans des centaines d'organismes différents, comme par exemple plasmodium malariae. Il s'agit des 2 séquences les plus courtes, d'une longueur de 6 aa (TNGTKR et HKNNKS). Les deux plus longues, (RSYLTPGDSSSG) et (QTNSPRRA), de 12 et 8 aa, sont effectivement uniques (ou quasi-uniques, RSYLTPGDSSSG se retrouve aussi dans flavobacterium luteum) à la fois au VIH1 et au SARS-Cov2. Dans le sida, la protéine gp120 joue un rôle équivalent à la protéine S en se liant au récepteur CD4 des lymphocytes T. La seule insertion non présente dans gp120 matche la protéine précurseur gag au niveau d'un site de clivage qui transposé dans le SARS-Cov2 constitue le site de la furine entre S1 et S2. Nous avons montré dans la partie 4 du chapitre précédente qu'elle importance revêtait ce site dans le caractère infectieux multi-organe du SARS-Cov2.
Finalement, Pradhan et al. analysent les positions relatives des trois autres insertions sur la structure spatiale du domaine S1 de la protéine S. Bien que situés à distance les uns des autres, sur la séquence linéaire du sous domaine N-terminal de S1 (SD-1), ces trois fragments de la protéine gp120 du VIH1 se trouvent regroupés à un endroit particulier (apex), bien exposé au cytoplasme et qui participe à la reconnaissance par contact du récepteur ACE2 de l'hôte. Ils concluent que l'élongation de séquences des boucles structurales de S1 au niveau de l'apex donne plus de flexibilité au domaine de liaison au récepteur ACE2. Vérifier la réalité de ces propos serait possible expérimentalement conjointement avec des simulations de dynamiques moléculaires. Il est indéniable que cette augmentation de la longueur de séquence favorise la flexibilité de la structure et pourrait donc, théoriquement, augmenter la capacité de cette région a engager des interactions favorables avec le récepteur ACE2 ou d'autres co-récepteurs associés. Le fait qu'il s'agisse de mini-séquences de la protéine gp120 revêt un intérêt particulier car elles possèdent, par tropisme d'évolution, des propriétés d'affinités spécifiques à certains récepteurs cellulaires.

La conclusion de Pradhan et al. est que cette conjonction spatiale d'insertions du VIH1, unique au SARS-Cov2, qui contribuerait à rendre le virus plus infectieux, en plus de l'insertion du site de clivage de la furine unique chez les betacoronavirus 2b, ne peut pas être fortuite. Cela sous-entendait qu'il s'agirait de modifications artificielles de gain de fonction (GOF), ce qui a conduit les auteurs à devoir rétracter leur article. Pourtant, on est bien obligé de constater, une fois de plus, qu'il y a quelque chose de troublant qui se surajoute à tout ce que nous avons décrit dans le chapitre précédent. Nous voyons ici également la proximité indéniable et unique de la protéine S du SARS-Cov2 avec celle du RaTG13.
Contre-expertise de l'analyse de Pradhan et al. indique la possibilité d'un tropisme naturel vers le VIH1.
Si Pradhan et al. avaient utilisé pour leur comparaison initiale le domaine S1-N-ter du SARS-Cov Urbani ils n'auraient pas pu identifier directement 4 points d'insertions mais seulement 2 (leur insert 3 et 4). En effet, au niveau des inserts 1 et 2 la séquence SARS-Cov Urbani a la même longueur (à un résidu près pour l'insert 2) et correspond également à des mini-séquences de la protéine gp120. Pour la position de l'insert 1, il s'agit de TNNAATKR que l'on retrouve 100% identiques dans les virus ZC45, ZXC21, et 86% identique dans le VIH1. Pour la position de l'insert 2, il s'agit de LSGYYHNNKT que l'on retrouve parmi les coronavirus uniquement dans les virus ZC45, ZXC21 et sous la forme LKGYFNNNKT dans le VIH1. Même si la séquence du SARS-Cov Urbani est plus courte de 3 résidus au niveau de l'insert 3 elle correspond exactement à une mini-séquence de la protéine gp120 (NNGWTA) qui se retrouve également uniquement dans les virus ZC45 (E0.002) et ZXC21(E0.035) et Pan-Cov-GD (E0.035) parmi tous les coronavirus.
Par acquis de conscience, nous avons analysé plus en détail la région de l'insertion 1 (encadrée par les triplets conservés SNT et YFA) dans une sélection de 20 betacoronavirus étalés dans le temps entre 2003 et 2019. Nous observons que la région de l'insertion 1 a une longueur très variable. Parmi les séquences analysées, 8 d'entre elles sont quasiment aussi longues que celle du SARS-Cov2. La plupart comportent également des fragments de séquences du VIH1 avec des valeurs d'espérance E < 14 (voir signification de E en annexe à cette partie) meilleures que celles correspondant au fragment VIH1 identifié dans le SARS-Cov2, en particulier chez les virus militaires ZC45 et ZXC21. Nous avons ensuite étudié le virus Shaanxi2011, un betacoronavirus de type SARS de chauve-souris, donc non humanisé.
La séquence complète QNTIVYFDNH de l'insert 1 est unique aux betacoronavirus Shaanxi2011, BtRs-BetaCoV/HuB2013 et au VIH1 (gp120) ainsi que 2 autres organismes. Nous retrouvons sur les boucles de la séquence du domaine S1 du Shaanxi2011, au niveau correspondant aux insertions 2 et 3, des mini-séquences du VIH1 (TVSRNQHY , E4.6, gag gene, protéine matrice p17a et QANFLTEN, E13 gag pol). La séquence TVSRNQHY se retrouve dans 2 autres betacoronavirus (BtRs-BetaCoV/HuB2013 et Cp/Yunnan2011) et dans 2 organismes autres que le VIH1. De même, la séquence QANFLTEN est unique aux betacoravirus Shaanxi2011 et BtRs-BetaCoV/HuB2013 parmi les coronavirus mais présente dans de multiples autres organismes.
Nous observons donc une situation similaire au SARS-Cov2 dans d'autres betacoronavirus comme le SARS-Cov Urbani (humain) et le Shaanxi2011 (chauve-souris), démontrant une relation naturelle distante entre les coronavirus et le VIH1.
La situation est moins flagrante pour le virus Shannxi2011 que pour le SARS-Cov2 et le SARS-Cov par ce que les séquences au niveau des insertions 2 et 3 ne correspondent pas à la protéine de pénétration cellulaire gp120. Pradhan et al. auraient dû pousser plus loin leur investigations. Étrange n'était pas le mot le plus approprié pour caractériser leurs observations mais malgré tout ils ont attiré l'attention sur un tropisme naturel, qui peut-être a fait l'objet de recherche GOF.
En résumé, c'est ce tropisme qui permettrait en particulier à la protéine S d'agir sur le récepteur cellulaire CD4 point de pénétration du VIH dans les lymphocytes T4, par l'intermédiaire des boucles rallongées et très flexibles de son domaine S1 qui s'apparentent par leur composition en acides aminés à celles de la protéine gp120. En effet, on observe des lymphopénies CD4 et CD8 et une exhaustion lymphocytaire chez les patients atteints des formes les plus graves de Covid-19. Les cellules T CD4+ expriment très peu le récepteur ACE2 indiquant que la protéine S se lie au récepteur CD4. Pour l'instant les études in vitro montrent que par chance le SARSCov2 ne peut pas se répliquer à l'intérieur des T CD4+ et CD8+ mais les observations cliniques montrent que des patients meurent de sepsis généralisées dues à un épuisement des lymphocytes T CD4+ et CD8+ comme dans le SIDA ...
Quel impact aux niveaux génomique et protéique des autres « insertions » du VIH1 repérées dans la protéine S du SARS-Cov2 ?
Nous revenons à présent sur les 16 « mini-séquences » apparentées au VIH (>80% d'identité) repérées par Luc Montagnier et Jean-Claude Perez au niveau du génome du virus SARS-Cov2. Comme nous l'avons vu précédemment, de telles séquences existent naturellement, réparties le long des génomes des coronavirus à SARS, comme le SARS-Cov, ce qui rend l'analyse de leur présence renforcée en certains endroits sujette à caution. D'ailleurs, ils précisent que 14 de ces 16 séquences étaient déjà présentes dans le SARS-Cov de 2002-2003. Le professeur Montagnier les appelle des éléments informatifs exogènes (EIE) car avec une longueur ≥ 18 nt elles pourraient avoir une signification biologiques en modifiant l'expression des gènes du SARS-Cov2. Ces EIE au nombre de 16 sont repérés dans l'Orf1a et le gène S du SARS-Cov2 et ce qui est très intriguant c'est que 8 d'entre eux soient concaténés à la queue-leu-leu sur une courte section de 275 nt au début du domaine S1-N-ter du gène S. Cela constitue le premier point clé des observations de Perez et Montagnier.


Le second point-clé soulevé par Perez et Montagnier est l'identification d'une séquence de 223 nt (21550 – 21772) commençant à l'intersection de l'Orf1ab et du gène S et chevauchant partiellement la région regroupant les 8 EIE (21694-21969). Selon eux, cette séquence est unique au SARS-Cov2 et au RaTG13 avec 91% d'identité de façon contiguë (sans insertions ou délétions) sur une longueur de 223 nt. Nous avons vérifié, pour nous en rendre compte nous-même, et nous trouvons que ce n'est pas tout à fait exact, car nous constatons aussi que le virus Pan-Cov-GX partage cette séquence unique avec 77% d'identité sur une longueur contiguë de 220 nt, et Pan-Cov-GD également sur 220 nt, avec des zones d'insertion. Aucun autre organisme répertorié dans GenBank ne partage cette séquence, ni de près ni de loin, mis à part les virus ZC45 et ZXC21 mais de façon très partielle sur une longueur d'environ150 nt seulement. Cela prouve une relation difficilement explicable entre ces 6 virus, séquencés dans un intervalle de temps de moins de deux ans. Une relation que nous avions également montré avec l'analyse de la région de Lyons Weiler présentée en partie 2 du chapitre 8.
Le troisième point clé est la séquence signature (TGTTTTTATTACTTTTATTGC CACTATTCTCT) de la souche Kénya 2008 du VIH1 qui se retrouve conservée à 91% dans le SARS-Cov2 et à 84% dans le RatG13, au tout début du gène S. La recherche inverse qui consiste à chercher les séquences proches de la séquence correspondante du SARS-Cov2 (TGTTTTTCTTGTTTTATTGCCACTATTCTCT) conduit à la même unicité de proximité. Ce genre de séquence signature est appelé aussi fingerprint car elle signe l'identité d'un organisme en étant unique à celui-ci. Il a plus de 18 milliards de milliards de combinaisons possibles de 32 nt. Cela ajouté au fait que les génomes ne sont pas organisés de façon aléatoire permet qu'une séquence aussi courte puisse identifier à coup sûr un organisme. Cette séquence appartient donc de façon unique au gène de la protéine vpu (une protéine nécessaire au relargage des virions par certaines cellules) de la souche Kénya 2008. Les seules séquences proches appartiennent à un papillon nocturne (Laspeyria flexula, 97% d'identité sur 28 nt) et une espèce de dauphin (Delphinapterus leucas, 93% d'identité sur 28 nt). Cela peut indiquer que ces organismes ont été contaminés dans un passé lointain par un virus parent de cette souche particulière du VIH1 dont des fragments sont été intégrés à leur génome.

Donc, la présence de la séquence signature traduit l'intégration d'un fragment de séquence de la souche Kénya 2008 au génome du SARS-Cov2... et à celui du RaTG13, dont le seul échantillon connu était conservé depuis 2013 (jusqu'en 2017-2018) dans de la paraffine à l'Institut de virologie de Wuhan. A moins d'imaginer que les chauves-souris chinoises du Yunnan aient été contaminées avant 2013 par un ancêtre de la souche Kénya 2008 du VIH1 et auraient produit naturellement une recombinaison virale on ne peut pas expliquer cela par le simple hasard. Une fois de plus cela signe la grande proximité difficilement explicable entre le SARS-Cov2 et le RaTG13. La question que l'on peut se poser est, au-delà du hasard d'une recombinaison, quel avantage fonctionnel aurait induit par recombinaison et sélection l'intégration d'une telle séquence dans les coronavirus à SARS ?
La présence de cette séquence semble d'autant plus inexplicable quelle correspond en fait au brin ARN négatif de la protéine vpu, par effet de symétrie comme dans un miroir. Au cours de la réplication d'un virus à ARN positif comme le SARS-Cov2, le brin négatif est créé par la polymérase du virus pour servir de matrice à la génération d'un nouveau brin positif qui sert à la transcription du gène dans les ribosomes. Donc, cette séquence ne code pas une séquence protéique de la protéine vpu du VIH1 dans le SARS-Cov2.
Dans le cas où il se serait agi du brin positif de la protéine vpu, une interprétation fonctionnelle était tout à fait possible car la vpu est une protéine accessoire importante pour la réplication du virus en favorisant entre autre le relargage des virions générés dans les cellules infectées. Donc, sur le plan GOF cette petite protéine a un grand intérêt car on note que sa « rétro » séquence partielle se trouve au tout début de la partie S1-N-ter du gène S, à l'extrême pointe de son apex qui constitue le bord d'attaque du virus sur la membrane cellulaire. En effet, sur une longueur de 18 aa, la partie N-ter de la protéine S flotte dans le cytoplasme et pourrait servir d'ancre pour s'arrimer sur la membrane.
La séquence brin positif (AGAGAATAGTGGCAATAAA AGTAATAAA AACA) se situe également à l'extrémité N-ter de la protéine vpu et correspond à une séquence très lipophile avec des charges positives (MVAIKVIKT), c'est-à-dire capable d'adhérer à la membrane cellulaire. Cependant, cette séquence particulière n'est pas transcrite en aa par ce que la vpu du VIH1 Kénya 2008 n'est pas fonctionnelle (non transcrite car son codon d'initiation est muté en ata). Une séquence N-ter d'une vpu fonctionnelle : MQPIPIVAIV montre un caractère lipophile semblable à celle du SARS-Cov2 S N-ter : MFVFLVLLPLV).

L'homologie brin négatif correspondrait-elle à un mécanisme de contrôle épigénétique dans l'hypothèse où le gène S serait intégré au génome humain par une rétro-transcriptase endogène ou exogène (voir chapitre 8 partie 1) ? Nous admettons notre grande ignorance en la matière et nous invitons les généticiens comme Alexandra Henrion Caude ou le professeur Montagnier à nous répondre.
Le quatrième point clé est la présence d'un longue séquence de nucléotides (41) du domaine S2 de la protéine S homologue (86% d'identité) à une séquence de Plasmodium Yoelii, l'agent du paludisme. Mais cette séquence se retrouve dans les virus Pan-Cov-GD (90% d'identité sur 41 nt) , RaTG13 (92% d'identité sur 39 nt), Pan-Cov-GX (90% d'identité sur 39 nt) et ZXC21 (90% d'identité sur 39 nt). Elle se retrouve également à hauteur de 85% sur 39 nt dans un grand nombre de coronavirus de type SARS, en particulier (ZC45, RsSHC014, Rs3367, ...). Elle appartient aussi au génome d'un papillon de nuit (Noctua fimbriata), du bourdon (Bombus Terrestris) et de la cigalle (Jikradia olitoria) prouvant que ces insectes sont en contact avec ce type de coronavirus ou le parasite Plasmodium Yoelii depuis un passé très lointain. Se pourrait-il que l'insertion de cette séquence dans le génome de ces insectes leur ait conféré un pouvoir de résistance au parasite Plasmodium Yoelii ?

Dans Yoelii, cette séquence correspond au brin positif codant pour une séquence de la protéine Fam-a dont la fonction n'est pas connue. Comme le cadre de lecture de cette séquence est décalé entre Yoelii et le SARS-Cov2 il n'y a pas d'identité direct de séquence au niveau aa. Dans Yoelii la séquence aa est KHKSNKFTKHKPKKN extrêmement riche en lysine K. Dans le SARS-Cov2 la séquence protéique correspondante est AQVKQIYKTPPIK insérée dans une plus longue séquence de 20 aa (KNTQEVFAQVKQIYKTPPIK) conservée à 100% entre le SARS-Cov2, le RaTG13, le Pan-Cov-GD et les virus ZC45 et ZXC21, mais aucune autre protéine de GenBank n'a cette séquence. Une fois de plus nous observons la relation du SARS-Cov2 avec les virus RaTG13 et Pan-Cov-GD, mais également ici avec les virus ZC45 et ZXC21. Une proximité que, par ailleurs, Li-Meng Yan clame haut et fort dans ses déclarations et ses articles (voir chapitre 8 partie 3).
Cette séquence se situe dans le peptide de fusion du domaine S2 de la protéine S. Le domaine S2 participe à la fusion du virus dans la membrane cellulaire dans un mécanisme complexe suite à des clivages protéolytiques. Dans l'assemblée supramoléculaire formée par le trimère S elle ne se retrouve pas directement exposée au cytoplasme, sauf après clivage par la furine. Est-il possible qu'elle puisse constituer à ce niveau un épitope vaccinal, cela paraît peu probable. Il faudrait que le peptide de fusion soit exposé au cytoplasme suffisamment longtemps et accessible aux anticorps. De plus le décalage du cadre de lecture empêche le codage d'une séquence aa similaire à Yoelii. Nous ne pouvons par en dire d'avantage.
Les mini-séquences du VIH1 et du VIH2 repérées par Perez et Montagnier sur une section courte de la protéine S peuvent-elles constituer des épitopes vaccinaux anti-VIH ?
Nous avons vu au début de cette partie qu'une approche vaccinale pouvait consister à insérer (ou échanger) des fragments de séquences virales, identifiées dans le virus ciblé comme pouvant générer des épitopes vaccinaux. Pour cela on utilise un support constitué par un autre virus, dont on maîtrise très bien la manipulation et l'inactivation, ce dont on doit avoir la certitude. Une autre stratégie, beaucoup plus récente, consiste à greffer une protéine enveloppe du virus cible (comme le VIH) sur une virus vecteur. On pourrait même pousser le concept de vaccination à un virus respiratoire, assez infectieux mais très peu pathogène, comme un coronavirus humain de rhume bénin, auquel la population est adaptée depuis longtemps et qui véhiculerait des épitopes vaccinaux du virus ou de l'agent pathogène cible. Nous ne savons pas si une telle stratégie est réalisable en pratique. En tout cas, vu ce qui a déjà été entrepris ces dernières années en matière de manipulations génétiques des virus, dans des buts louables en Chine et ailleurs, ce n'est certainement pas une règle morale qui empêcherait d'envisager sérieusement cette possibilité.
Les nombreux travaux de recherche conduits depuis une douzaine d'années à l'Institut de virologie de Wuhan (WIV) dénotent des efforts considérables qui y ont été déployés pour trouver un vaccin contre le sida. Ce vaccin passe par l'identification d'épitopes vaccinaux conservés au travers de la multitude de variants d'une même souche et d'épitopes capables de générer des anticorps à large spectre, c'est-à-dire capable de reconnaître de multiple épitopes. Une condition essentielle pour qu'une séquence d'acides aminés puisse devenir un épitope est qu'elle soit exposée en surface de la protéine et accessible par les anticorps.
Analyse des EIE sous l'angle d'épitopes vaccinaux possibles.
Lorsque nous évaluons la possibilité que les 8 EIE du point-clé 2 constituent des épitopes vaccinaux nous nous heurtons au même problème que pour le point-clé numéro 3, c'est-à- dire que 5 mini-séquences sur 8 correspondent au brin négatif de l'ARN des virus VIH1 et VIH2. Ce sont les séquences repérées dans l'ordre 1, 5, 6, 7 et 8. Donc, elle ne peuvent en aucun être transcrites en acides aminés pouvant générer une réponse immunitaire. On pourrait imaginer une manipulation subtile du code ARN brin positif de sorte que le brin négatif devienne codant. En effet, on observe sur le gène S à la fin de la section Lyons-Weiler la présence de la séquence cat (gène S :1196-1198) suivi 60 nt plus loin par la séquence attata (gène S : 1259 – 1264) qui rendraient le brin négatif codant avec le promoteur de transcription tataat suivi du codon initiation atg. Cependant, cette idée se heurte à la présence de tta (S : 1216) qui serait transcrit en codon stop (taa).
Sur les trois EIE correspondant au brin positif codant du VIH/SIV (séquence 2,3 et 4), la séquence 4 bien que correspondant à un match sur brin positif est décalée d'un nucléotide sur le plan translationnel et donc ne code pas les mêmes acides aminés ce qui l'exclut comme épitope possible.
Finalement, les EIE 2 (CTGGGACTAATGGTACTAAG) et 3 (AATGGTACTAAAAGGTTAGATAACACTG) correspondent réellement à des séquences d'acides aminés homologues du SIV et du VIH1. Les séquences aa 2 (SGTNGTK) et 3 (NGTKRFDNPV) se chevauchent en recouvrant l'insertion 1 de Pradhan et al. C'est une région complètement exposée au cytoplasme et donc candidate idéale pour constituer des épitopes vaccinaux. Étant donné l'hyper-variabilité de la protéine gp120 dont l'homologie de séquence est très basse, à la fois au sein des virus VIH1 (50%) et entre le VIH1 et le SIV (40%), il est difficile de déterminer à priori avec certitude la position exacte de la séquence GTNGTK de l'EIE par rapport à une quelconque structure X-ray de la protéine gp120. Cependant, il semble bien qu'il s'agisse de la boucle S22-S23, particulièrement exposée au cytoplasme et située à un endroit qui empêcherait, si il était bloqué par un anticorps, l'accrochage du récepteur CD4. Celle de l'EIE 3 correspond également sans aucun doute à la boucle S17-S18 de la gp120 du VIH1.

Nous voyons donc qu'effectivement deux des EIE correspondent parfaitement à la définition d'épitopes vaccinaux pour des souches de VIH1 et le SIV. Cependant, ils ne permettraient pas d'élaborer un vaccin car ils sont situés sur les boucles hypervariables de la protéine gp120. Nous trouvons des situations identiques avec les virus Pan-Cov GD (boucle S16-S17 de la protéine S), Pan-Cov-GX (boucle S17-S18) et RsSHC014 (boucle S20-S21).
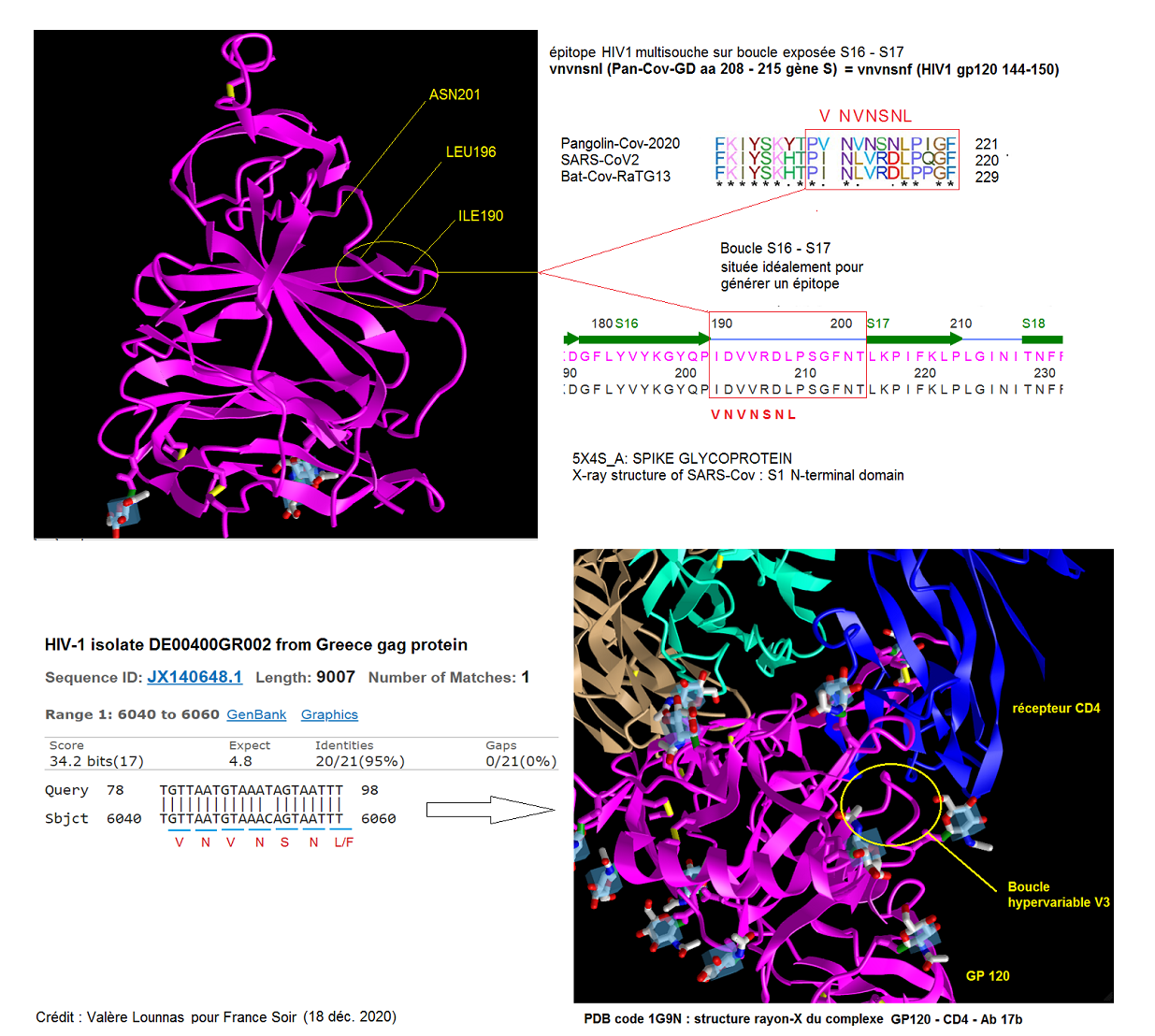
En conclusion, étant donné que seuls 2 EIE sur 8 pourraient correspondre à des épitopes vaccinaux, et que ce serait de toute façon une stratégie vouée à l'échec (en raison de l'hyper-variabilité de la protéine gp120), nous sommes obligés d'abandonner l'idée d'une cassette d'épitopes vaccinaux classique pour expliquer le présence très concentrée de ces mini-séquences VIH dans une section du domaine S1 de la protéine S. La raison de cette présence statistiquement improbable reste donc inexpliquée et est à chercher ailleurs.
Il est étrange de constater que cette région très restreinte est soumise à une très grande pression évolutive positive par rapport au reste du génome du SARS-Cov2. Comme nous l'avions expliqué dans la partie 3 du chapitre 8, cela ne peut traduire qu'un besoin d'adaptation à l'hôte (qui aurait dû se produire au tout début de l'épidémie). Cela pourrait aussi correspondre à une nécessité d'harmonisation globale du génome ce qui pourrait assurément signer une incohérence due à une insertions récente avant le déclenchement de l'épidémie (possibilité de manipulation génétique). La protéine S du virus RaTG13, conservé dans un échantillon paraffiné avant d'être séquencé entre 2017 et 2018, aurait-elle été branchée sur un virus cousin du RaTG13 ?

Dans la partie suivante, nous aborderons cet aspect méconnu de la génomique mise en évidence par la méthode de résonance fractale de Jean-Claude Perez basée sur la suite mathématique de Fibonacci. Cette approche permet également d'établir le degré d'adaptation des virus à leurs hôtes.
document annexe qui explique la signification des alignements de séquence
Rédaction achevée le 14 février 2021
À LIRE AUSSI


L'article vous a plu ? Il a mobilisé notre rédaction qui ne vit que de vos dons.
L'information a un coût, d'autant plus que la concurrence des rédactions subventionnées impose un surcroît de rigueur et de professionnalisme.
Avec votre soutien, France-Soir continuera à proposer ses articles gratuitement car nous pensons que tout le monde doit avoir accès à une information libre et indépendante pour se forger sa propre opinion.
Vous êtes la condition sine qua non à notre existence, soutenez-nous pour que France-Soir demeure le média français qui fait s’exprimer les plus légitimes.
Si vous le pouvez, soutenez-nous mensuellement, à partir de seulement 1€. Votre impact en faveur d’une presse libre n’en sera que plus fort. Merci.

Tous les vendredis, recevez la newsletter hebdo de France-Soir
